Abstract
Breast cancer is a highly heterogeneous disease, with tumors capable of adapting to shifting conditions, making the development of effective personalized therapies particularly challenging. Patient-derived models, such as patient-derived organoids (PDOs) and circulating tumor cell (CTC) cultures, have emerged as powerful tools for investigating intra- and inter-tumor heterogeneity. These models largely retain the genetic, phenotypic, and microenvironmental features of the original tumors, providing valuable insights into disease progression, drug response, and resistance mechanisms. Furthermore, by enabling tumors’ spatiotemporal molecular profiling, PDOs and CTCs offer a dynamic approach to assess treatment efficacy over time. However, to fully capture the complexity of breast cancer heterogeneity, it is required to develop models from multiple tumor and blood samples collected throughout the course of treatment. This review explores the potential of integrating PDOs and CTC models to better understand intra-tumor heterogeneity while addressing key challenges in developing patient-derived models that accurately recapitulate patients’ tumors to advance personalized care. The integration of PDOs and CTCs could represent a paradigm shift in the personalized management of metastatic breast cancer.
1. Introduction
Breast cancer remains the most frequently diagnosed cancer and the second leading cause of cancer-related deaths among women [1]. While advances in early-stage adjuvant therapies and targeted treatments for metastatic breast cancer have significantly improved patient survival, the development of treatment resistance remains a significant challenge, leading to incurable disease in the metastatic setting [2,3]. Breast cancer classification into clinical subtypes is crucial for guiding treatment selection, but high molecular diversity within each breast cancer subtype, defined as inter-tumor heterogeneity, significantly contributes to treatment resistance [4,5]. Furthermore, there is significant variability in cancer cells within a single tumor or patient, known as intra-tumor heterogeneity [6,7]. In metastatic disease, more aggressive cell populations can emerge in different metastatic sites, adapt to distinct microenvironments, and spread to new locations [8,9,10]. Targeted therapies can exacerbate this heterogeneity by selecting resistant cell clones [11]. Furthermore, immune-related events within the tumor microenvironment (TME), such as the diversity of tumor-infiltrating lymphocytes (TILs) and the expression of programmed cell death protein-1 (PD-1) and ligand-1 (PD-L1), can vary across the different metastasized organs within a patient. This variability may contribute to distinct responses to immune checkpoint inhibitors in different metastatic sites [12].
Currently, tumor histopathological analysis and molecular testing often rely on a single biopsy, which fails to capture the full genetic diversity within solid tumors, as tumors exhibit significant genetic heterogeneity across different regions and metastatic sites. Additionally, the evolving nature of tumors over time makes a single biopsy insufficient to represent the dynamic genetic landscape (Figure 1). Therefore, obtaining samples from multiple metastatic sites and longitudinal blood or tumor samples is crucial for comprehensively analyzing the tumor’s genetic profile and selecting more effective, personalized treatment strategies.
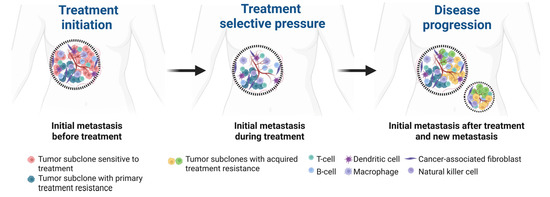
Figure 1.
Spatial and temporal heterogeneity of metastatic breast cancer. The initial metastatic site harbors a subclone sensitive to therapy (red) and a second subclone with primary treatment resistance (blue). Under treatment selective pressure, new subclones with acquired resistance and enhanced metastatic potential (green and yellow) emerge within the initial metastatic site and subsequently disseminate to establish secondary metastatic sites. Created in BioRender.com.
Patient-derived organoids (PDOs) are three-dimensional (3D) cellular structures that can self-renew and differentiate and, when developed from different regions of the primary tumor and multiple metastatic sites, enable studying single-cell heterogeneity and functional diversity within a single patient. Moreover, circulating tumor cells (CTCs) obtained from liquid biopsies offer a non-invasive approach to monitor real-time treatment responses and assess tumors’ genomic, transcriptomic, and proteomic diversities [13,14,15]. This review explores the opportunities and challenges of the complementary use of matched patient-specific PDOs and CTCs to study tumor heterogeneity and treatment resistance in advanced breast cancer.
2. Tumor Heterogeneity in Metastatic Breast Cancer
Breast cancer is a highly heterogeneous disease characterized by diverse histopathological and molecular subtypes that can evolve, both as the disease progresses and after treatment-induced selective pressure. The dynamic spatial and temporal diversity within an individual patient’s tumor (intra-tumor heterogeneity) and across different patients with the same cancer subtype (inter-tumor heterogeneity) contribute to distinct treatment responses and the development of treatment resistance, either intrinsic (preexisting resistance mechanisms) or acquired (resistance mechanisms emerging during treatment) [16,17]. Thus, developing successful personalized therapies requires a deeper understanding of tumor heterogeneity and access to preclinical models that recapitulate this complexity.
Breast cancer is traditionally classified by immunohistochemistry into three clinical subtypes: hormone receptor-positive (HR+)/human epidermal growth factor receptor 2 negative (HER2−), HER2+, and triple-negative breast cancer (TNBC). This classification remains the one in use to guide current therapeutic strategies for breast cancer, which provides only a broad measure of inter-tumor heterogeneity and results in insufficiently tailored treatments [4]. Molecular profiling has further refined breast cancer subtypes into four main groups, luminal HR+ A and B, HER2-enriched, and basal-like, while also identifying distinct transcriptomic subgroups within TNBC [18]. Indeed, more than 20 distinct subtypes that differ genetically, morphologically, and clinically, and over 1600 driver mutations have been identified in breast cancer, highlighting its heterogeneous nature [19,20]. Nevertheless, bulk tissue molecular profiling used in these studies has limitations in assessing intra-tumor heterogeneity [12]. Single-cell analysis is needed to understand tumor genomic and transcriptomic instability, which drive the selection and outgrowth of diverse cancer clones, leading to various cellular phenotypes and treatment resistance mechanisms [21,22].
Tumor heterogeneity is also influenced by diverse non-tumor components constituting the TME, such as immune cells, fibroblasts, endothelial cells, adipose tissue, soluble signaling molecules, and extracellular matrix ECM [23]. These interact with cancer cells by secreting and expressing factors that promote tumor growth by suppressing anti-tumor immune response, or block tumor growth by activating anti-tumor immunity [24,25]. The TME varies across breast cancer subtypes in both the quantity and composition of immune cell populations [26]. TNBC has been considered the most “immune-hot” subtype, but HER2-amplified and high-grade HR+ luminal B tumors may also exhibit significant numbers of TILs and increased immune checkpoint expression. In contrast, luminal A tumors have shown to be largely immune-cold [27,28]. An unmet need in cancer research remains the effective capture of tumor and TME heterogeneity in preclinical models that can be functionally tested in vitro. Standardized and reproducible models that reflect intra- and inter-tumor heterogeneity are critical for advancing precision oncology and developing effective personalized therapies.
3. Capturing Tumor Heterogeneity with In Vitro Patient-Derived Models
3.1. Overview of PDOs in Cancer Research
PDOs are a self-organized, 3D assembly of cancer cells derived directly from a patient’s tumor that have emerged as a promising and cost-effective tool for modeling cancer heterogeneity. PDOs can be composed of tumor cells alone or incorporate the TME, such as immune and stromal cells, either in a native or reconstituted model [29]. PDOs can recapitulate many of the structural, molecular, and cellular features of the original tumor, including tumor subtype markers, mutations, copy number alterations, and cellular architecture. Because PDOs maintain, at least initially, the heterogeneity present in the original tumors, they serve as an excellent tool for studying both inter-tumor and intra-tumor heterogeneity [30]. These models represent a substantial improvement compared to other and more used human-derived breast cancer models, including cancer cell lines and patient-derived xenografts (PDXs), whose development is inefficient, labor-intensive, and time-consuming, making their widespread use for preclinical testing impractical. Furthermore, cell lines and PDXs are typically generated from advanced-stage tumors, limiting their ability to fully represent the cancer spectrum [31]. In breast cancer, PDOs have been shown to preserve the histological and molecular characteristics of the original tumors, including key markers such as estrogen receptor (ER), progesterone receptor (PR), and HER2, copy number variations, and sequence alterations [29]. Notably, breast cancer organoids have been successfully developed across all major gene expression-based subtypes and enable in vitro drug screening, with results that align with in vivo xenotransplantation findings and patient responses [29]. An overview of PDOs used as preclinical models for breast cancer is shown in Table 1.

Table 1.
Selected studies on breast cancer PDOs and their applications.
3.1.1. PDO Culture Methods
Significant progress has been made in recent years in the methodologies for deriving cancer organoids. Advances in tissue sourcing now allow organoid generation from a range of specimens, including surgical resections, core needle biopsies, liquid effusions, and even blood samples. While it is possible to establish PDOs from tumor cells isolated from blood and effusion samples, this review focuses on PDOs derived from solid tumors, which remain the most widely used source. The most commonly utilized cancer organoid model is the submerged culture system, wherein mechanically and enzymatically dissociated tumor cells are embedded in an ECM and cultured under a medium enriched with growth factors and pathway regulators tailored to the specific tumor type [38,39,40]. A key modification in breast cancer organoid culture protocols has been the addition of Neuregulin 1, a ligand for HER3 and HER4 involved in mammary development and tumorigenesis, which enables efficient organoid generation and long-term expansion [31]. The addition of a Rho-associated protein kinase (ROCK) inhibitor further enhanced culture conditions by promoting the survival and proliferation of tumor epithelial cells. Media optimization tests further revealed that Wnt-3A had minimal impact, epidermal growth factor (EGF) concentrations above 5 ng/mL increased proliferation but disrupted organoid integrity, and addition of a p38 MAPK inhibitor reduced organoid establishment efficiency at concentrations above 1 μM [31].
While the submerged culture method enables efficient organoid generation and easy scalability, it is limited in its ability to maintain non-epithelial cells from the TME. To address this limitation, alternative methods have been developed, including the air–liquid interface (ALI) culture system, where minced tumor fragments are directly embedded in collagen within a transwell, with the upper layer exposed to air while the lower layer is in contact with culture media [40,41]. Another method involves first culturing minced tumor tissue in low-attachment plates to form spheroids, which are then transferred to a microfluidic device with controlled flow and regulated concentrations of gel and media through microchannels to provide a stable and dynamic environment for the spheroids [42,43]. Both ALI and microfluidic systems allow studying immunotherapy responses and characterizing immune cell populations within the TME [44,45,46]. The most advanced approach constitutes the organ-on-a-chip system, which integrates microfluidics, tissue engineering, and cell biology to simulate organ-level human physiology [47]. This approach allows precise control of nutrient and signaling gradients and can model complex tissue environments by incorporating multiple cell types, but its high cost, complexity, and need for specialized training and equipment limit widespread use.
3.1.2. Co-Culture Models of Tumor and Tumor Microenvironment
A significant advancement of PDO technology is its ability to model the TME in a patient-specific manner, accurately replicating both the tumor architecture and immune and non-immune composition. This is crucial for testing the growing number of therapies targeting the TME. The TME is a complex and dynamic network of immune, stromal, and endothelial cells, adipose tissue, ECM components, and soluble factors that regulate tumor progression, metastasis, and response to therapy [48,49]. Including immune and stromal cells into PDOs through a reconstituted or native model enables a comprehensive study of TME–tumor interactions and their impact on tumor behavior and treatment outcomes.
The conventional submerged organoid models are easier to generate from cancer tissue, but their lack of stromal components halts the study of TME-targeted therapies. More complex organoid models that incorporate both tumor and non-tumor components have been developed, either by reconstitution of isolated epithelial, stromal, and immune cells in already developed tumor PDOs (bottom-up reconstitution) or by maintaining the native architecture of the tumor by simply mincing the tumor tissue without enzymatic digestion (top-down reconstitution, Figure 2).
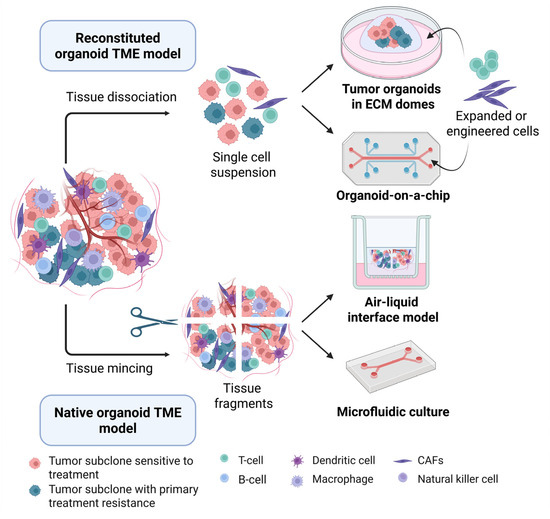
Figure 2.
Development of tumor organoid culture systems to model the TME. Reconstituted organoid TME models (top), including submerged cultures and organoids-on-a-chip, are derived from tumor single-cell suspensions and subsequently co-cultured with selected cell populations of interest, such as engineered or isolated and expanded immune and stromal cells, to precisely control the composition of the microenvironment. Native tumor TME models (bottom) are generated by physically mincing or gently dissociating tumor tissue into fragments, then culturing the fragments in an air-liquid interface or microfluidic culture system, which preserve endogenous immune and stromal cells for a limited period. Abbreviations: CAFs, cancer-associated fibroblasts; ECM, extracellular matrix; and TME, tumor microenvironment. Created in BioRender.com.
Reconstituted TME organoid models are developed by first generating single-cell suspensions from enzymatically and mechanically digested tumor tissue. These tumor cells are initially grown in an ECM and then co-cultured with engineered and/or previously expanded immune cells, such as TILs or autologous peripheral blood immune cells used to expand tumor-reactive T cell clones [50,51]. Monocyte-derived macrophages, have also been successfully co-cultured with human intestinal organoids in chip-based models of inflammatory bowel disease [52]. Tumor organoids and immune cell co-cultures are also promising models for testing the killing capacity of engineered immunotherapies, such as chimeric antigen receptor (CAR) T cells, in solid tumors [53,54]. In breast cancer, 3D organotypic co-culture systems combining natural killer (NK) cells with tumor organoids have been developed to model tumor–immune cell interactions and to test NK cell-mediated cytotoxicity against metastatic breast cancer cells [37]. Another study using organ-on-a-chip technology developed a tri-culture system, including a dual-compartment chip to investigate tumor matrix remodeling, a 3D tubular chip to study bone metastasis using bone marrow-derived stem cells and endothelial cells, and a liver–tumor co-culture chip that mimics hepatic metabolism for drug screening [55]. In contrast to this reconstituted model that establishes tumor PDOs before adding TME components, co-cultures can also be derived directly after enzymatic tumor digestion with non-purified tumor cell suspensions for short-term co-culture of tumor and non-tumor cells [56,57]. While this model retains the patient TME cell populations, dissociation into single cells disrupts the tumor and TME architecture and spatial conformation and may alter crucial cell interactions between tumor and non-tumor cells that affect tumor progression and treatment response [58,59]. Indeed, the new spatial distribution following dissociation into single-cell suspensions may completely remove some tumor–TME functional interactions and introduce new interactions between cell types that were previously separated, as well as expose tumor and non-tumor cells to non-physiological ECM, which can change cancer and non-cancer cell morphology and function [29].
Other approaches, such as the ALI model and microfluidic devices, are used to study the native tumor and TME within their original architecture. In contrast to reconstituted models, these organoid systems allows the growth of tumor cells en bloc alongside endogenous non-tumor cells, retaining both the heterogeneity of cell populations, such as cancer-associated fibroblasts (CAFs), cytotoxic T lymphocytes (CTLs), T-helper cells, tumor-associated macrophages (TAMs), NK cells, and B cells and the tumor architecture and spatial arrangement of its components [42,43,60].
The accurate representation of the 3D tumor architecture, heterogeneity, and treatment response by PDOs containing TME presents an excellent opportunity to test patient-specific responses to immunotherapy. PDOs reconstituted with peripheral blood lymphocytes or TILs have been used to investigate patients’ responses to checkpoint blockade or adoptive cell therapy with variable success [53,61,62,63,64]. Another study explored the role of CAFs in influencing pancreatic ductal adenocarcinoma (PDAC) organoid behavior. This study identified two distinct CAF subtypes, myofibroblastic and immunoinflammatory, that interacted differently with the organoids, highlighting CAF subtype heterogeneity and its impact on tumor progression and therapy response [65]. Notably, CAF-secreted interleukin-6 promotes epithelial–mesenchymal transition (EMT) and drug resistance in esophageal adenocarcinoma, suggesting these as potential biomarkers for improved patient stratification [66]. In breast cancer, a whole-tumor cell (WTC) ex vivo model was successfully established from 98 out of 116 tumor samples, effectively recapitulating the parental tumor architecture and endogenous microenvironment. This model demonstrated strong clinical correlations and predictive value across a wide range of breast cancer therapies [35].
Noteworthily, the utility of TME organoid models for testing immunotherapy response and resistance alongside clinical treatments is limited by their short viability, potentially missing treatment resistance driven by rare tumor clones. Comparing patient-matched PDOs from pre- and post-treatment samples and developing immune organoid models from both primary and metastatic tumors could improve the study of immunotherapy efficacy in late-stage cancers. Furthermore, reconstituted models that incorporate only a single immune subtype may require greater complexity by the addition of several key TME components to improve both short- and long-term predictive accuracy. Lastly, advancements in 3D culture techniques, such as ECM scaffolds, microfluidics, and media optimization, could further optimize and standardize TME organoid models [67].
3.1.3. Applications of PDOs in Drug Testing and Response Prediction
PDOs are a promising tool for drug screening and predicting therapeutic responses. Studies have shown that drug testing in PDOs generally aligns with patient responses, though most investigations have been limited to small cohorts [30]. Although more extensive studies remain scarce, a study with a pancreatic cancer PDO biobank showed an 89% match between PDOs and patient responses [68]. Another study with gastrointestinal tumor organoids successfully predicted outcomes in 20 of 21 cases [69]. Specifically in breast cancer, 12 PDOs were established from needle biopsies of metastatic lesions, and two models, tamoxifen-sensitive and -resistant, were selected for in vitro and in vivo testing. The observed drug responses closely mirrored those of the corresponding patients, demonstrating clinical relevance [31]. Similarly, another study generated PDOs from eight breast cancer patients, including diverse molecular subtypes and cases of multidrug resistance. These PDOs demonstrated drug response profiles that were consistent with patient clinical outcomes, revealing either sensitivity or resistance to a wide range of therapies, including epirubicin, paclitaxel, docetaxel, cyclophosphamide, gemcitabine, vinorelbine, trastuzumab, pyrotinib, olaparib, 5-fluorouridine, etoposide, and alpelisib [32]. These findings highlight the potential of PDOs to inform and guide personalized treatment decisions. Furthermore, PDOs are useful for drug resistance mechanisms studies by evaluating resistant clones, which may harbor mutations or altered signaling pathways that help them evade therapy. Identifying these pathways can inform the development of combination therapies, where one drug targets a key pathway central for the resistant clones while another targets central pathways for both resistant and sensitive cells. PDOs also provide an excellent platform for studying tumor metastasis. By exposing PDOs to different treatment regimens and monitoring their growth and survival, researchers can better understand the dynamics of metastatic spread and the role of intra-tumor heterogeneity in facilitating metastasis. These studies help elucidate how specific clones within a tumor are better equipped to survive in different microenvironments, which is crucial for developing more effective treatments that target metastatic disease.
However, organoids can change in culture, as subclonal mutations may be gained or lost over passages, while truncal mutations generally remain stable [70,71]. Indeed, early-passage organoids closely reflect the mutational profile of the original tumor tissue, but genetic drift and treatment selection pressure in culture often lead to divergence over time [29]. Moreover, gene expression and epigenetic alterations may be influenced by culture conditions, which do not fully replicate the in vivo environment [29]. Furthermore, organoids represent only the tumor sample from which they were derived, limiting their ability to capture the full spectrum of the tumor heterogeneity. A more comprehensive approach requires culturing samples from multiple tumor regions and distinct metastatic sites.
Importantly, organoids derived from different metastatic sites of a patient with pancreatic cancer showed comparable responses to some chemotherapies but varied significantly with others [68]. Similarly, PDOs developed from different regions of the same colorectal tumor exhibited up to a 30-fold variation in treatment responses, underscoring the need for multiple sampling to capture intra-tumor heterogeneity fully [72]. Notably, a study analyzing organoids from 27 samples across five liver cancer patients found relatively low variability in drug response between patients but significant differences among samples from the same patient [29]. Additionally, a large colorectal cancer study involving multi-omic analysis of 78 clonally derived organoids from multiple tumor regions of three patients revealed substantial variability in mutations, gene expression, epigenetic modifications, and drug responses among clonal organoids from the same region [73]. Together, these studies highlight the potential of PDOs in capturing key aspects of tumor heterogeneity and predicting treatment responses but underscore the need for sampling multiple tumor regions and metastatic sites to represent intra-tumor heterogeneity accurately. Furthermore, the lack of standardization in organoid development/culture methodologies remains a major source of variability, impairing reproducibility and comparability across studies and limiting their clinical application [29]. Implementing PDOs in real-world clinical settings faces additional challenges, including the time-consuming process of establishing and testing organoids that may delay treatment decisions, the high costs associated with specialized laboratory infrastructure and skilled personnel, and logistical difficulties in promptly acquiring and processing fresh tumor samples. Together, these limitations hinder the broad clinical use of PDO-based approaches.
3.2. Overview of CTCs in Metastatic Cancer
CTCs are cancer cells shed from the primary tumor or metastasis into the bloodstream and play a key role in the metastatic process by intravasation and extravasation from the circulation and colonization of distant organs [74,75] (Figure 3). CTCs are extremely rare, with numbers in peripheral blood of about 1 CTC per 1–10 million white blood cells [76]. In contrast to tumor resections and biopsies, CTCs can be collected non-invasively and repeatedly, enabling longitudinal monitoring of spatial and temporal changes in the tumor’s genetic and phenotypic profiles throughout disease progression and treatment response [77]. Thus, CTCs hold exceptional potential in prognosis prediction, real-time monitoring of treatment response, and personalized drug susceptibility testing [78].
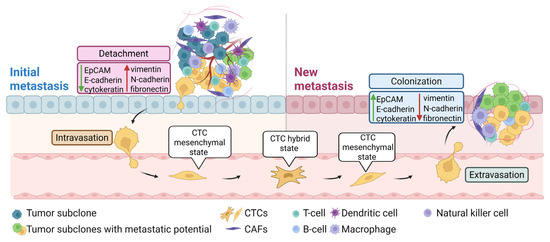
Figure 3.
CTCs and EMT in the metastatic process. Tumor cells at the initial metastatic site undergo EMT to invade surrounding tissue, enabling their entry into the bloodstream (intravasation). This process involves upregulation of mesenchymal markers, such as vimentin, N-cadherin, and fibronectin and downregulation of epithelial markers, such as EpCAM, E-cadherin, and cytokeratin. In the bloodstream, CTCs adopt a partial or hybrid epithelial–mesenchymal state, enhancing their metastatic potential. Upon reaching a distant site, mesenchymal CTCs exit the bloodstream (extravasation), invade the new tissue, and revert to an epithelial state to establish a metastatic colony. Abbreviations: CAFs, cancer-associated fibroblasts; CTCs, circulating tumor cells; and EMT, epithelial–mesenchymal transition. Created in BioRender.com.
CTCs exhibit high heterogeneity and a wide range of molecular alterations, including mutations, gene expression alterations, and epigenetic modifications, contributing to their ability to survive in circulation and colonize distant organs. Despite their heterogeneity, all metastasis-competent CTCs can self-replicate similarly to stem cells [79]. To maintain a balance between migration and proliferation, metastasis-competent CTCs do not become completely mesenchymal but acquire sufficient mesenchymal features, defined as a partial or hybrid EMT state [80]. This plastic and transient state enables cells to co-express both epithelial and mesenchymal markers, enhancing their tumorigenicity, stemness, metastatic potential, and therapy resistance [81].
3.2.1. CTCs Isolation
In contrast to other liquid biopsy components, such as circulating tumor DNA (ctDNA) and tumor-derived extracellular vesicles (tdEVs), CTCs are a “complete” biological material that can be used to isolate DNA, RNA, and protein simultaneously. Furthermore, in vitro culture of isolated CTCs, although challenging, enables functional studies on metastasis development and drug testing. However, the isolation of CTCs poses a significant challenge due to their rarity and heterogeneity. Most separation techniques rely on either the physical properties, such as size and deformability, or biological characteristics, such as surface markers, of CTCs (Figure 4). Advances in microfluidics, size-based filtration, and immunomagnetic capture have improved the efficiency and reliability of CTC isolation from patient blood samples, but no current method can consistently isolate a pure population of CTCs [74,82,83]. Limitations such as low recovery rates, poor purity, and reduced cell viability, continue to restrict the expansion of CTCs in vitro or in vivo, limiting their use in downstream functional analyses [84].
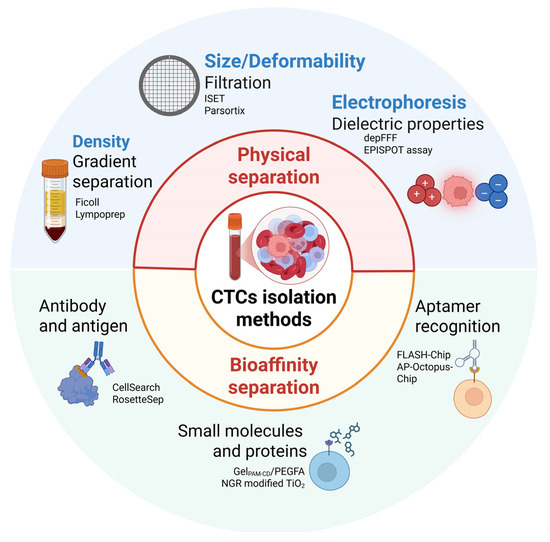
Figure 4.
Overview of CTC isolation methods. CTC isolation and enrichment methods are based on either physical properties, such as size, density and dielectric characteristics, or immunoaffinity approaches, which rely on specific binding to antibodies, small molecules, or aptamers. Abbreviations: AP-Octopus-Chip, aptamer-functionalized octopus chip; CTCs, circulating tumor cells; depFFF, dielectric electrophoresis field flow fractionation; FLASH-Chip, fluidic multivalent grafted nanointerface microfluidic chip; GelPAM-CD, β-cyclodextrin (β-CD)-functionalized poly(acrylamide) hydrogel; ISET, isolating by size of epithelial tumor cells; PEGFA, polyethylene glycol folic acid; and NGR, asparagine-glycine-arginine peptide. Created in BioRender.com.
CTC isolation methods based on physical properties take advantage of distinct characteristics of these cells, including their larger size, greater stiffness, unique density, and dielectric properties compared to other blood cells [75,85]. Size-based enrichment approaches, including filtration methods, separate CTCs, typically 20–30 μm in diameter, from smaller blood cells (8–12 μm). Filtration-based approaches are simple, cost-effective, and high-throughput, but due to heterogeneity of the size and deformability among CTCs, smaller or more flexible tumor cells may be lost, making it difficult to define optimal thresholds. Common size-based systems include ISET (Isolation by Size of Epithelial Tumor Cells), ScreenCell, and the FDA-cleared Parsortix system, which combines microfluidics and stepped physical structures to enhance capture efficiency [86,87,88]. Other physical-based methods use density differences between CTCs and blood cells for separation by density gradient centrifugation with media such as Lymphoprep or Ficoll-Paque. This method enables simple and rapid enrichment of label-free, viable CTCs. However, due to overlapping densities between CTCs and mononuclear cells, the yield of highly pure CTCs is low, often requiring additional purification steps [89]. Moreover, CTCs exhibit distinct dielectric properties compared to other peripheral blood cells, and thus, when exposed to alternating electric fields, their polarization behavior differs, enabling spatial separation from other cells [90]. An example of this approach is dielectric electrophoresis field-flow fractionation (depFFF), which enables label-free CTC isolation based on dielectric differences [91].
Bioaffinity-based separation methods utilize immunoreactions between antibodies, aptamers, peptides, or small molecules and target proteins or receptors on the surface of the CTCs to enable their recognition, enrichment, and isolation [90]. Positive selection with antibodies against EpCAM or cytokeratins, or negative selection of leucocytes with anti-cluster of differentiation CD45 antibody, followed by separation of antibody-tagged cells with magnetic beads, are commonly used to separate viable CTCs [75]. The FDA-approved assay CellSearch identifies CTCs based on cytokeratin expression, lack of CD45, and nuclear labeling with DAPI [75]. Furthermore, this method enables the collection of CTC-clusters, tdEVs, and apoptotic bodies, and the antibodies used can be replaced by antibodies targeting other antigens of interest. Similarly, the CellCollector allows the detection and isolation ex vivo of EpCAM-expressing cells [92]. Noteworthy, selection based on EpCAM and cytokeratin levels may only capture a subset of CTCs, as these may exhibit a highly variable expression of epithelial markers. Indeed, CTCs with mesenchymal or basal-like phenotypes may exhibit extremely low levels of epithelial markers [93,94]. Furthermore, cells processed with CellSearch must be permeabilized before staining with antibodies, limiting the downstream applications of the isolated CTCs. As a result, antigen-agnostic methods such as Parsortix remain valuable, as these allow the separation of CTCs based on size and mechanical properties, such as deformability, hereby facilitating downstream expansion both in vitro and in vivo [95,96,97]. Notably, xenografting of CTCs directly after isolation or following expansion in vitro has also been performed to further expand the CTC numbers [98,99]. Breast cancer CTCs have been subcutaneously injected into the mammary fat pad of SCID mice and successfully formed tumors expressing hormone receptors that matched the patients’ tumors [100,101]. Interestingly, it has been shown that breast cancer CTC lines expanded in vivo were capable of metastasizing at the same sites as in the patients the CTCs were isolated from [102]. Currently, 37 breast cancer cell lines derived from CTCs have been reported, of which 11 cell lines were cultured long-term for over 6 months [100,102,103,104,105,106,107].
In addition to EpCAM and cytokeratin, various unique surface proteins expressed on CTCs can be targeted to distinguish them from other cells. By exploiting the specific binding interactions between these proteins and their ligands or substrates, CTCs can be selectively captured and subsequently released [90]. An example is the folate receptor (FR), a cysteine-rich cell surface glycoprotein that binds folic acid (FA) with high affinity, which is minimally expressed in most normal tissues but overexpressed in many cancers to support rapid cell division under low folate conditions. This makes FR a useful target for CTC enrichment and isolation, and has been used to develop a β-cyclodextrin-functionalized poly(acrylamide) hydrogel (GelPAM-CD) with FA conjugated to adamantane-PEG, forming the final scaffold GelPAM-CD/PEGFA for selective CTC capture [108]. Similarly, the small-molecule peptide NGR (asparagine-glycine-arginine) targets aminopeptidase N (CD13), a tumor cell membrane protein, enabling CTC capture in blood [109]. Additionally, aptamers, short DNA or RNA sequences that fold into specific structures, offer another approach to target molecules on the CTC surface. Compared to antibodies, aptamers have advantages including high affinity, small size, low cost, ease of synthesis, and precise target recognition. Furthermore, unlike antibodies, aptamers enable gentle CTC release by temperature or hybridization-induced conformational changes, preserving cell integrity post-capture. These properties have been used to design devices such as the fluidic multivalent grafted nanointerface microfluidic chip (FLASH-Chip) and the aptamer-functionalized octopus chip (AP-Octopus-Chip) [110,111]. While physical and bioaffinity-based methods capture CTCs, detection rates vary due to differing identification criteria. Indeed, the FDA-approved CellSearch system correlates CTC count with prognosis but is limited clinically because its reliance on EpCAM+, CK+, DAPI+, CD45− markers may miss CTCs undergoing EMT, which often show reduced EpCAM and CK expression. To overcome these limitations, integrated microfluidic chips combining multiple detection methods are being developed to enable high-efficiency capture, intact protein profiling, compatibility with single-cell sequencing, and live-cell drug testing. Additionally, because CTC counts in small blood samples are low, in vivo enrichment technologies are emerging to sample broader circulatory volumes, enhancing CTC yield for downstream analyses.
3.2.2. CTCs Prognostic and Predictive Potential in Breast Cancer
The FDA-approved CellSearch system has supported CTCs as a strong and independent prognostic biomarker for early and metastatic breast cancer [112]. In the neoadjuvant setting, the prognostic significance of CTCs detected by CellSearch was evaluated in the GeparQuattro trial in patients with operable or locally advanced HER2+ breast cancer, both before and after neoadjuvant therapy with HER2-targeted therapy together with anthracycline-taxane-based chemotherapy. Shorter disease-free survival (DFS) and overall survival (OS) were observed for ≥1 or ≥2 CTCs/7.5 mL before neoadjuvant chemotherapy, while no significant association was found with the CTC numbers after neoadjuvant chemotherapy [113]. In the adjuvant setting, the presence of CTCs was assessed with CellSearch before and two years after chemotherapy in early-stage, high-risk breast cancer patients enrolled in the phase III SUCCESS A trial [114]. This trial compared two adjuvant chemotherapy regimens followed by either two or five years of zoledronate. Detection of CTCs two years post-chemotherapy was independently associated with poor OS and DFS, regardless of CTC status at baseline. Notably, patients who were CTC-positive both at baseline and at the two-year follow-up showed the poorest survival outcomes [114]. However, undetectable levels of CTCs for most early-stage breast cancer patients hinders the clinical utility of CTCs in this setting, and for those with detectable levels, the low numbers complicate molecular testing [115]. Nevertheless, baseline CTC detection remains a strong predictor of poor prognosis, supporting treatment escalation in high-risk groups. Noteworthily, since not all CTCs possess metastatic potential, and given the fact that those that do may not survive in the bloodstream, CTC detection does not necessarily indicate poor clinical outcomes [116]. In the metastatic setting, a prospective multicenter study evaluated 177 patients with measurable metastatic breast cancer for CTC levels using CellSearch, both before starting a new line of treatment (baseline) and at the first follow-up visit. CTC levels at both time points independently predicted progression-free survival (PFS) and OS, independently of the time to metastasis, site of metastasis, or HRstatus. Importantly, CTC levels predicted poor OS and PFS earlier than traditional imaging methods, with changes detectable three to four weeks after therapy initiation compared to eight to 12 weeks by imaging [82]. Additionally, CTC dynamics after treatment can inform prognosis, as patients with high baseline CTC counts but rapidly decreasing numbers during treatment have a favorable prognosis [117]. However, despite their prognostic potential, CTCs are not yet a validated standard for guiding therapeutic decisions due to insufficient standardization.
Beyond their prognostic value, CTC enumeration and surface marker expression have also been explored for their predictive potential [118]. The STIC CTC study, a multicenter randomized phase III trial, compared first-line therapy (ET or chemotherapy) based on CTC count (CTC arm) versus the investigator’s choice (standard arm). The trial showed that the CTC-guided approach was noninferior to clinician-driven treatment in terms of 2-year PFS. While no OS benefit was observed in the general population, patients with a high CTC count but low clinical risk experienced a significant OS advantage from chemotherapy, highlighting the potential of CTC counts to guide the choice between ET and chemotherapy [118,119,120].
Notably, several studies have investigated whether HER2+ CTCs in HER2- metastatic breast cancer could predict response to HER2-targeted therapy. A multicenter phase II trial evaluated the efficacy of lapatinib in metastatic breast cancer patients whose primary tumors were HER2- but had HER2+ CTCs, as isolated by CellSearch and assessed for HER2 status by immunofluorescence (IF) [121]. CTC positivity was defined as ≥2 CTC per 7.5 mL of blood, and HER2 positivity as ≥50% of the CTCs expressing HER2. Among the 139 HER2- patients screened, 96 patients were CTC+ (median number of CTCs: 19; range 2–1637) and seven of these (7%) had HER2+ CTCs and were eligible for treatment with lapatinib. No objective tumor responses were observed although one patient achieved disease stabilization for 8.5 months. Similarly, the DETECT III trial investigated the benefit of HER2-targeted therapy in HER2- metastatic breast cancer patients with HER2+ CTCs [122]. Of 2137 patients screened, 101 were eligible and randomized to receive standard therapy with or without lapatinib. The primary endpoint was CTC clearance (absence of CTCs at the end of treatment), while secondary endpoints included PFS, OS, and safety. Although CTC clearance rates did not significantly differ between groups, patients treated with lapatinib had an improved OS compared to the standard therapy group (20.5 vs. 9.1 months, p = 0.009).
The CirCe01 trial assessed the clinical utility of CTC-based monitoring in metastatic breast cancer patients beyond the third-line of chemotherapy. Patients with ≥5 CTC per 7.5 mL, as determined by CellSearch, were randomized to standard arm or CTC-arm, where CTC counts were evaluated after each chemotherapy cycle, and early tumor progression, predicted by rising CTCs, led to switching to subsequent therapies. The trial found no difference in OS between the two arms and failed to demonstrate the clinical utility of CTC monitoring [121]. Overall, trials evaluating the predictive value of CTCs have shown inconsistent results, and further and more robust studies are needed to establish their definitive clinical role [120,121,122,123]. An overview of these studies is shown in Table 2.
Table 2.
Clinical studies using CTCs detected by CellSearch technology for prognostic or predictive applications in breast cancer.
3.2.3. CTCs Molecular and Functional Analysis
Once isolated, CTCs can undergo various analyses beyond enumeration, including molecular profiling techniques, such as single-cell RNA and DNA sequencing, to gain insights into their genetic and phenotypic profiles. Whole-genome sequencing of CTCs can identify mutations shared with the primary tumor, as well as novel subclonal mutations that may drive metastasis and treatment resistance. However, due to the low number of CTCs, cell expansion is necessary for functional studies. These studies may help to assess the most aggressive, metastatic cells, those with self-renewal capacity in culture, while also evaluating their molecular profiles [92]. Additionally, the ex vivo culture of CTCs may be used to assess the efficacy of specific treatments and investigate the development of drug resistance mechanisms [124]. Advances in microfluid chips have allowed the quick process of whole blood samples for simultaneous CTC isolation and drug testing [125]. Indeed, CTC cultures derived from patients with metastatic ER+ breast cancer were tested by next-generation sequencing and with a panel of targeted inhibitors, including PI3K, FGF, IGF, and ER targeting drugs, as well as cytotoxic agents, including doxorubicin, paclitaxel, and capecitabine [100]. Notably, the CTC line exhibiting a PIK3CA mutation showed high sensitivity towards the PI3Kα isoform-specific inhibitor BYL719 in vitro and in vivo [100]. In another study, the cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor palbociclib was tested in a CTC cell line compared to MCF7 cells, which showed sensitivity of CTCs to this drug, supporting CDK4/6 inhibitor clinical benefit for this patient [102].
Multi-modal single-cell assays can capture the complete CTC profile, including its genome, transcriptome, proteome, and epigenome. Additionally, multiparametric analysis of various liquid biopsy components, such as ctDNA and tdEVs, can provide complementary tumor information, particularly in early-stage disease, where CTC numbers are low. The short lifespan of CTCs in circulation suggests continuous renewal and release, whereas ctDNA primarily reflects dead tumor cells and passive cellular debris shedding. As a result, ctDNA is better suited for detecting minimal residual disease and identifying gene alterations than CTCs [126].
4. Integrating Patient-Derived Models from Solid and Liquid Biopsies to Study Tumor Heterogeneity in Metastatic Breast Cancer
4.1. Opportunities for Combined Use of PDOs and CTCs
While tumor and liquid biopsies have been extensively molecularly profiled independently, their integration offers a more comprehensive view of tumor heterogeneity in metastatic breast cancer. Integrating functional drug testing using PDOs generated from tumor samples with real-time genomic monitoring of CTCs isolated from liquid biopsies could enable dynamic and personalized treatment strategies. Importantly, concurrent analysis of both solid and liquid biopsies may be required to detect variants missed by either method alone [127]. Studies have shown that over half of the mutations detected in either tissue or liquid biopsy are not common across biopsies, highlighting their complementary potential [128,129]. In fact, the addition of plasma testing to tissue increases therapeutic target detection for patients with metastatic non-small-cell lung cancer by 15% [130]. Moreover, concordance among frequent alterations varies across genes and decreases with longer time intervals between tissue and blood collection [131]. Specifically, ESR1 variants are more likely to be detected in circulating cell-free DNA (cfDNA) than tissue, whereas certain variants in PIK3CA are more commonly identified in tissue samples, suggesting that a combined approach improves detection of alterations [127]. Indeed, while driver events are frequently shared between multiple biopsies, acquired alterations detected in liquid biopsies are frequently polyclonal, present at low allelic fractions, and are indicative of multi-clonal convergent evolution. Furthermore, significant genomic variability has also been observed across primary tumors, relapses, and metastatic biopsies in patients with endocrine therapy resistance [132]. This intra-tumor heterogeneity poses clinical challenges for selecting optimal treatments and identifying resistance mechanisms. Therefore, integrating matched patient-derived models from serial solid and liquid biopsies offers a promising strategy to capture and understand the spatiotemporal dynamics of intra-tumor heterogeneity.
Individually, both PDOs and CTCs offer unique insights into tumor behavior (Table 3). PDOs model the 3D architecture and genetic landscape of the tumor, while CTCs allow for real-time assessment of metastatic potential and the clonal diversity of cancer cells circulating in the bloodstream. Integrating these two models enables a holistic view of tumor heterogeneity and evolution, providing a platform to monitor how breast cancer adapts under therapeutic pressure (Figure 5). Notably, pairing PDOs with CTC analyses from the same patient enables tracking of the tumor’s spatial and temporal clonal evolution, offering insights into the adaptive strategies that cancer cells use to survive treatment. Growing evidence supports the concept of metastatic plasticity in breast cancer, including clinical reports of atypical metastatic patterns, such as duodenal involvement, underscoring the need for patient-derived models capable of capturing both clonal diversity and spatial heterogeneity [133]. The combined use of PDOs and CTCs may provide a more accurate representation of metastatic behavior in real-time, supporting more precise therapeutic decision-making. The emergence of treatment resistance mechanisms in PDOs can be compared to the corresponding CTC profiles to pinpoint which clones demonstrate resistance and invasion abilities and how these clones respond to subsequent treatment cycles. This cross-model tracking allows mapping the clonal selection and adaptation process under therapeutic pressure, providing valuable data on the persistence of resistant cell populations and the likelihood of specific clones contributing to disease progression.
Table 3.
Overview of the main features of PDOs and CTCs as preclinical cancer models.
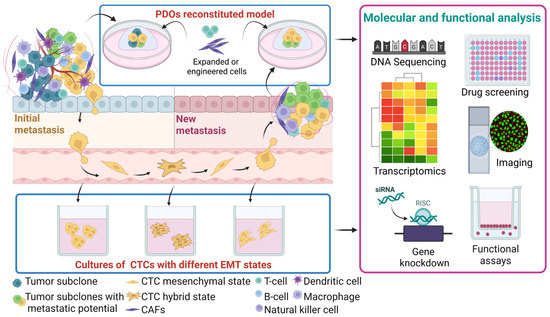
Figure 5.
Development of patient-matched PDOs and CTCs to decipher spatiotemporal intra-tumor heterogeneity. PDOs developed from the initial and secondary metastatic sites enable molecular and functional profiling of tumor heterogeneity during disease progression. This approach is complemented by the parallel characterization of matched CTC cultures, providing insights into the most aggressive tumor cell states. Comparative analyses among the initial tumor, disseminated CTCs, and new metastatic lesions provide a dynamic view of tumor evolution. Abbreviations: CAFs, cancer-associated fibroblasts; CTCs, circulating tumor cells; EMT, epithelial–mesenchymal transition; and PDOs, patient-derived organoids. Created in BioRender.com.
This complementary approach may also help identify new molecular targets for combination therapies by finding specific gene expression patterns or resistance markers shared across PDOs and CTCs. Testing new drug combinations on resistant PDO clones and profiling CTCs for survival markers may contribute to more robust and personalized treatment strategies. In addition, by identifying and targeting resistant clones early, it may be possible to reduce relapse rates and improve long-term outcomes in breast cancer. This approach enhances the predictive power of preclinical models and identifies potential therapeutic targets that might otherwise be overlooked when using PDOs or CTCs separately.
As the field of precision oncology advances, the combined use of PDOs and CTCs can become an integral part of clinical decision-making, particularly in the metastatic setting. This dual-model approach offers a rapid, iterative method to test and refine treatment regimens based on both initial tumor-derived organoid characteristics and real-time responses in CTCs. The adaptability of these models to incorporate new genetic information or emerging resistance mechanisms may enable clinicians to adjust treatments proactively, thus likely improving patient outcomes and reducing resistance-driven progression. Moreover, as larger datasets from PDO and CTC studies become available, these insights can be leveraged to develop predictive algorithms, enabling oncologists to choose the most effective treatment regimen for the individual patient.
4.2. Limitations and Future Directions
PDOs and CTCs are undeniably crucial tools for translational cancer research, but significant challenges regarding their use remain. In the case of PDOs, these are difficult to maintain long-term, and their growth may favor specific clones, leading to potential bias in drug testing. Although early passage PDOs often maintain the genetic composition of the original tumor, the extent of clonal selection and genetic drift in later passage PDOs, as well as following freezing-thawing cycles, might be a challenge for some tumors [30,134]. Another major limitation of traditional PDOs is the lack of key tumor-associated components, such as immune cells and fibroblasts, highlighting the need to develop organoid models that more faithfully replicate the TME to advance precision cancer therapy [30]. This variability is further influenced by the complex media composition required for the growth of PDOs. The use of conditioned media containing factors such as Wnt3a, R-spondin, and Noggin and the presence of serum can impede long-term culture viability due to their complexity and cost-effectiveness [135]. Importantly, a key challenge is the lack of standardization of organoid media across different laboratory batches, which may lead to selective pressure favoring specific cell populations and biased results. Recently developed, advanced artificial Wnt agonists may offer promising, cost-effective, standardized, serum-free alternatives to previous Wnt-conditioned media [136,137]. Regarding CTCs, these are rare cells that are difficult to isolate in sufficient quantities for comprehensive analysis and require special culture conditions that are difficult to replicate in vitro. Furthermore, only a small subset of CTCs in the bloodstream may contribute to metastasis formation.
Various studies have shown the potential of PDO and CTC functional drug testing for predicting patient responses to treatment, though more extensive prospective validation is still required. Importantly, these assays should be complemented with clinical measurements of survival outcomes. The establishment of PDO and CTC biobanks representing various cancer subtypes could accelerate drug screening and facilitate the identification of therapeutic response patterns in patients with similar tumor molecular profiles. Furthermore, genome-editing techniques could enable genome-wide functional studies, expand target screening, and enhance small molecule testing beyond traditional drug panels, thereby broadening the range of treatments tested [138,139,140,141]. Scaling up PDOs and CTC cultures in ultra-high-throughput systems would further enhance the feasibility of both genetic and drug testing approaches.
5. Conclusions
PDOs and CTCs are revolutionizing the study of breast tumor heterogeneity. These models offer unique insights into genetic, morphological, and microenvironmental diversity that underly cancer progression, metastasis, and treatment resistance. By integrating advanced molecular profiling of PDOs and CTCs, it will be possible to achieve a deeper understanding of the clonal evolution of breast cancer and develop more effective and personalized treatment strategies. Although further clinical validation is needed, PDOs and CTCs represent key tools for the effective implementation of precision oncology in advanced breast cancer.
Author Contributions
C.L.A. wrote the first draft. B.P. and N.N. modified and optimized it subsequently. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by grants from Pink Tribute (10205), the A.P. Møller foundation (2024-00976) and the Danish Cancer Research Foundation (FID2628418).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ALI | air-liquid interface |
| AP-Octopus-Chip | aptamer-functionalized octopus chip |
| CAFs | cancer-associated fibroblasts |
| CAR T | chimeric antigen receptor T cells |
| CDK | cyclin-dependent kinase |
| CTCs | circulating tumor cells |
| cfDNA | circulating cell-free DNA |
| ctDNA | circulating tumor DNA |
| CTLs | cytotoxic T lymphocytes |
| depFFF | dielectric electrophoresis field flow fractionation |
| DFS | disease-free survival |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EMT | epithelial–mesenchymal transition |
| ER | estrogen receptor |
| FA | folic acid |
| FLASH-Chip | fluidic multivalent grafted nanointerface microfluidic chip |
| FR | folate receptor |
| GelPAM-CD | β-cyclodextrin (β-CD)-functionalized poly(acrylamide) hydrogel |
| HER2 | human epidermal growth factor receptor 2 |
| HR | hormone receptor |
| IF | immunofluorescence |
| IHC | immunohistochemistry |
| ISET | isolating by size of epithelial tumor cells |
| NGR | asparagine-glycine-arginine peptide |
| NK | natural killer cells |
| OS | overall survival |
| PD-1 | programmed cell death protein-1 |
| PD-L1 | programmed cell death protein-1 ligand |
| PDAC | pancreatic ductal adenocarcinoma |
| PDOs | patient-derived organoids |
| PDXs | patient-derived xenografts |
| PEGFA | polyethylene glycol folic acid |
| PFS | progression-free survival |
| PR | progesterone receptor |
| RNA-seq | RNA-sequencing |
| ROCK | Rho-associated protein kinase |
| TAMs | tumor-associated macrophages |
| tdEVs | tumor-derived extracellular vesicles |
| TILs | tumor-infiltrating lymphocytes |
| TME | tumor microenvironment |
| TNBC | triple-negative breast cancer |
| 3D | three-dimensional |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Mandelblatt, J.S.; Kurian, A.W. Change in Survival in Metastatic Breast Cancer with Treatment Advances: Meta-Analysis and Systematic Review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef] [PubMed]
- Agostinetto, E.; Gligorov, J.; Piccart, M. Systemic therapy for early-stage breast cancer: Learning from the past to build the future. Nat. Rev. Clin. Oncol. 2022, 19, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.H.; Ellis, I.; Allison, K.; Brogi, E.; Fox, S.B.; Lakhani, S.; Lazar, A.J.; Morris, E.A.; Sahin, A.; Salgado, R.; et al. The 2019 World Health Organization classification of tumours of the breast. Histopathology 2020, 77, 181–185. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. S2), S26–S35. [Google Scholar] [CrossRef]
- Koren, S.; Bentires-Alj, M. Breast Tumor Heterogeneity: Source of Fitness, Hurdle for Therapy. Mol. Cell 2015, 60, 537–546. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Ullah, I.; Karthik, G.M.; Alkodsi, A.; Kjällquist, U.; Stålhammar, G.; Lövrot, J.; Martinez, N.F.; Lagergren, J.; Hautaniemi, S.; Hartman, J.; et al. Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J. Clin. Investig. 2018, 128, 1355–1370. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothé, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Pasha, N.; Turner, N.C. Understanding and overcoming tumor heterogeneity in metastatic breast cancer treatment. Nat. Cancer 2021, 2, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Mannavola, F.; Pellè, E.; Quaresmini, D.; Tucci, M.; Silvestris, F. Liquid biopsy of cancer: A multimodal diagnostic tool in clinical oncology. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R.; et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef]
- Kwan, T.T.; Bardia, A.; Spring, L.M.; Giobbie-Hurder, A.; Kalinich, M.; Dubash, T.; Sundaresan, T.; Hong, X.; LiCausi, J.A.; Ho, U.; et al. A Digital RNA Signature of Circulating Tumor Cells Predicting Early Therapeutic Response in Localized and Metastatic Breast Cancer. Cancer Discov. 2018, 8, 1286–1299. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Bernard, E.; Abbosh, C.; André, F.; Auwerx, J.; Balmain, A.; Bar-Sagi, D.; Bernards, R.; Bullman, S.; DeGregori, J.; et al. Embracing cancer complexity: Hallmarks of systemic disease. Cell 2024, 187, 1589–1616. [Google Scholar] [CrossRef] [PubMed]
- Runa, F.; Hamalian, S.; Meade, K.; Shisgal, P.; Gray, P.C.; Kelber, J.A. Tumor microenvironment heterogeneity: Challenges and opportunities. Curr. Mol. Biol. Rep. 2017, 3, 218–229. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Danenberg, E.; Bardwell, H.; Zanotelli, V.R.T.; Provenzano, E.; Chin, S.F.; Rueda, O.M.; Green, A.; Rakha, E.; Aparicio, S.; Ellis, I.O.; et al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat. Genet. 2022, 54, 660–669. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef]
- Harris, M.A.; Savas, P.; Virassamy, B.; O’Malley, M.M.R.; Kay, J.; Mueller, S.N.; Mackay, L.K.; Salgado, R.; Loi, S. Towards targeting the breast cancer immune microenvironment. Nat. Rev. Cancer 2024, 24, 554–577. [Google Scholar] [CrossRef]
- LeSavage, B.L.; Suhar, R.A.; Broguiere, N.; Lutolf, M.P.; Heilshorn, S.C. Next-generation cancer organoids. Nat. Mater. 2022, 21, 143–159. [Google Scholar] [CrossRef]
- Lo, Y.H.; Karlsson, K.; Kuo, C.J. Applications of Organoids for Cancer Biology and Precision Medicine. Nat. Cancer 2020, 1, 761–773. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e310. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, W.; Zhang, Y.; Chen, Y.; Shan, C.; Li, J.; Jia, Y.; Li, C.; Du, C.; Cai, Y.; et al. Establishment of patient-derived organoids for guiding personalized therapies in breast cancer patients. Int. J. Cancer 2024, 155, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, X.; Ding, R.; Yang, L.; Lyu, X.; Zeng, J.; Lei, J.H.; Wang, L.; Bi, J.; Shao, N.; et al. Patient-Derived Organoids Can Guide Personalized-Therapies for Patients with Advanced Breast Cancer. Adv. Sci. 2021, 8, e2101176. [Google Scholar] [CrossRef]
- Goldhammer, N.; Kim, J.; Timmermans-Wielenga, V.; Petersen, O.W. Characterization of organoid cultured human breast cancer. Breast Cancer Res. 2019, 21, 141. [Google Scholar] [CrossRef]
- Chen, X.; Sifakis, E.G.; Robertson, S.; Neo, S.Y.; Jun, S.H.; Tong, L.; Hui Min, A.T.; Lövrot, J.; Hellgren, R.; Margolin, S.; et al. Breast cancer patient-derived whole-tumor cell culture model for efficient drug profiling and treatment response prediction. Proc. Natl. Acad. Sci. USA 2023, 120, e2209856120. [Google Scholar] [CrossRef]
- Campaner, E.; Zannini, A.; Santorsola, M.; Bonazza, D.; Bottin, C.; Cancila, V.; Tripodo, C.; Bortul, M.; Zanconati, F.; Schoeftner, S.; et al. Breast Cancer Organoids Model Patient-Specific Response to Drug Treatment. Cancers 2020, 12, 3869. [Google Scholar] [CrossRef]
- Chan, I.S.; Ewald, A.J. Organoid Co-culture Methods to Capture Cancer Cell-Natural Killer Cell Interactions. Methods Mol. Biol. 2022, 2463, 235–250. [Google Scholar]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab. Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef]
- Lai, B.F.L.; Lu, R.X.Z.; Davenport Huyer, L.; Kakinoki, S.; Yazbeck, J.; Wang, E.Y.; Wu, Q.; Zhang, B.; Radisic, M. A well plate-based multiplexed platform for incorporation of organoids into an organ-on-a-chip system with a perfusable vasculature. Nat. Protoc. 2021, 16, 2158–2189. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e1512. [Google Scholar] [CrossRef]
- Zhou, G.; Lieshout, R.; van Tienderen, G.S.; de Ruiter, V.; van Royen, M.E.; Boor, P.P.C.; Magré, L.; Desai, J.; Köten, K.; Kan, Y.Y.; et al. Modelling immune cytotoxicity for cholangiocarcinoma with tumour-derived organoids and effector T cells. Br. J. Cancer 2022, 127, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Beaurivage, C.; Kanapeckaite, A.; Loomans, C.; Erdmann, K.S.; Stallen, J.; Janssen, R.A.J. Development of a human primary gut-on-a-chip to model inflammatory processes. Sci. Rep. 2020, 10, 21475. [Google Scholar] [CrossRef] [PubMed]
- Schnalzger, T.E.; de Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Alieva, M.; Cleven, A.; Keramati, F.; Wezenaar, A.K.L.; van Vliet, E.J.; Puschhof, J.; Brazda, P.; Johanna, I.; Meringa, A.D.; et al. Uncovering the mode of action of engineered T cells in patient cancer organoids. Nat. Biotechnol. 2023, 41, 60–69. [Google Scholar] [CrossRef]
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2014, 35, 2454–2461. [Google Scholar] [CrossRef]
- Ding, S.; Hsu, C.; Wang, Z.; Natesh, N.R.; Millen, R.; Negrete, M.; Giroux, N.; Rivera, G.O.; Dohlman, A.; Bose, S.; et al. Patient-derived micro-organospheres enable clinical precision oncology. Cell Stem Cell 2022, 29, 905–917.e906. [Google Scholar] [CrossRef]
- Ou, L.; Liu, S.; Wang, H.; Guo, Y.; Guan, L.; Shen, L.; Luo, R.; Elder, D.E.; Huang, A.C.; Karakousis, G.; et al. Patient-derived melanoma organoid models facilitate the assessment of immunotherapies. eBioMedicine 2023, 92, 104614. [Google Scholar] [CrossRef]
- Schürch, C.M.; Bhate, S.S.; Barlow, G.L.; Phillips, D.J.; Noti, L.; Zlobec, I.; Chu, P.; Black, S.; Demeter, J.; McIlwain, D.R.; et al. Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 2020, 182, 1341–1359.e1319. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e422. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e1916. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.C.H.; Guerra, G.R.; Millen, R.M.; Roth, S.; Xu, H.; Neeson, P.J.; Darcy, P.K.; Kershaw, M.H.; Sampurno, S.; Malaterre, J.; et al. Tumor-Infiltrating Lymphocyte Function Predicts Response to Neoadjuvant Chemoradiotherapy in Locally Advanced Rectal Cancer. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Votanopoulos, K.I.; Forsythe, S.; Sivakumar, H.; Mazzocchi, A.; Aleman, J.; Miller, L.; Levine, E.; Triozzi, P.; Skardal, A. Model of Patient-Specific Immune-Enhanced Organoids for Immunotherapy Screening: Feasibility Study. Ann. Surg. Oncol. 2020, 27, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Exposito, R.; Semiannikova, M.; Griffiths, B.; Khan, K.; Barber, L.J.; Woolston, A.; Spain, G.; von Loga, K.; Challoner, B.; Patel, R.; et al. CEA expression heterogeneity and plasticity confer resistance to the CEA-targeting bispecific immunotherapy antibody cibisatamab (CEA-TCB) in patient-derived colorectal cancer organoids. J. Immunother. Cancer 2019, 7, 101. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef]
- Polak, R.; Zhang, E.T.; Kuo, C.J. Cancer organoids 2.0: Modelling the complexity of the tumour immune microenvironment. Nat. Rev. Cancer 2024, 24, 523–539. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e517. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Andrieux, G.; Boehnke, K.; Keil, M.; Silvestri, A.; Silvestrov, M.; Keilholz, U.; Haybaeck, J.; Erdmann, G.; Sachse, C.; et al. Heterogeneous pathway activation and drug response modelled in colorectal-tumor-derived 3D cultures. PLoS Genet. 2019, 15, e1008076. [Google Scholar] [CrossRef]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef]
- Dive, C.; Brady, G. SnapShot: Circulating Tumor Cells. Cell 2017, 168, 742–742.e741. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Genna, A.; Vanwynsberghe, A.M.; Villard, A.V.; Pottier, C.; Ancel, J.; Polette, M.; Gilles, C. EMT-Associated Heterogeneity in Circulating Tumor Cells: Sticky Friends on the Road to Metastasis. Cancers 2020, 12, 1632. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Hernández, L.E.; Eslami-S, Z.; Pantel, K.; Alix-Panabières, C. Molecular and Functional Characterization of Circulating Tumor Cells: From Discovery to Clinical Application. Clin. Chem. 2019, 66, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Eslami, S.Z.; Cortés-Hernández, L.E.; Thomas, F.; Pantel, K.; Alix-Panabières, C. Functional analysis of circulating tumour cells: The KEY to understand the biology of the metastatic cascade. Br. J. Cancer 2022, 127, 800–810. [Google Scholar] [CrossRef]
- Parkinson, D.R.; Dracopoli, N.; Petty, B.G.; Compton, C.; Cristofanilli, M.; Deisseroth, A.; Hayes, D.F.; Kapke, G.; Kumar, P.; Lee, J.; et al. Considerations in the development of circulating tumor cell technology for clinical use. J. Transl. Med. 2012, 10, 138. [Google Scholar] [CrossRef]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.; Schütze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M.; et al. Isolation by size of epithelial tumor cells: A new method for the immunomorphological and molecular characterization of circulatingtumor cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Desitter, I.; Guerrouahen, B.S.; Benali-Furet, N.; Wechsler, J.; Jänne, P.A.; Kuang, Y.; Yanagita, M.; Wang, L.; Berkowitz, J.A.; Distel, R.J.; et al. A new device for rapid isolation by size and characterization of rare circulating tumor cells. Anticancer. Res. 2011, 31, 427–441. [Google Scholar]
- Hvichia, G.E.; Parveen, Z.; Wagner, C.; Janning, M.; Quidde, J.; Stein, A.; Müller, V.; Loges, S.; Neves, R.P.; Stoecklein, N.H.; et al. A novel microfluidic platform for size and deformability based separation and the subsequent molecular characterization of viable circulating tumor cells. Int. J. Cancer 2016, 138, 2894–2904. [Google Scholar] [CrossRef]
- Lin, M.X.; Hyun, K.A.; Moon, H.S.; Sim, T.S.; Lee, J.G.; Park, J.C.; Lee, S.S.; Jung, H.I. Continuous labeling of circulating tumor cells with microbeads using a vortex micromixer for highly selective isolation. Biosens. Bioelectron. 2013, 40, 63–67. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, B.; Cai, J.; Yang, Y.; Tang, C.; Zheng, X.; Li, H.; Xu, F. Enrichment and separation technology for evaluation of circulating tumor cells. Talanta 2025, 282, 127025. [Google Scholar] [CrossRef]
- Gascoyne, P.R.; Noshari, J.; Anderson, T.J.; Becker, F.F. Isolation of rare cells from cell mixtures by dielectrophoresis. Electrophoresis 2009, 30, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Saucedo-Zeni, N.; Mewes, S.; Niestroj, R.; Gasiorowski, L.; Murawa, D.; Nowaczyk, P.; Tomasi, T.; Weber, E.; Dworacki, G.; Morgenthaler, N.G.; et al. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Int. J. Oncol. 2012, 41, 1241–1250. [Google Scholar]
- He, Y.; Shi, J.; Shi, G.; Xu, X.; Liu, Q.; Liu, C.; Gao, Z.; Bai, J.; Shan, B. Using the New CellCollector to Capture Circulating Tumor Cells from Blood in Different Groups of Pulmonary Disease: A Cohort Study. Sci. Rep. 2017, 7, 9542. [Google Scholar] [CrossRef] [PubMed]
- Peeters, D.J.E.; van Dam, P.J.; Van den Eynden, G.G.M.; Rutten, A.; Wuyts, H.; Pouillon, L.; Peeters, M.; Pauwels, P.; Van Laere, S.J.; van Dam, P.A.; et al. Detection and prognostic significance of circulating tumour cells in patients with metastatic breast cancer according to immunohistochemical subtypes. Br. J. Cancer 2014, 110, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Sequist, L.V.; Nagrath, S.; Toner, M.; Haber, D.A.; Lynch, T.J. The CTC-chip: An exciting new tool to detect circulating tumor cells in lung cancer patients. J. Thorac. Oncol. 2009, 4, 281–283. [Google Scholar] [CrossRef]
- Miller, M.C.; Robinson, P.S.; Wagner, C.; O’Shannessy, D.J. The Parsortix™ Cell Separation System-A versatile liquid biopsy platform. Cytom. A 2018, 93, 1234–1239. [Google Scholar] [CrossRef]
- Wang, R.; Chu, G.C.Y.; Mrdenovic, S.; Annamalai, A.A.; Hendifar, A.E.; Nissen, N.N.; Tomlinson, J.S.; Lewis, M.; Palanisamy, N.; Tseng, H.R.; et al. Cultured circulating tumor cells and their derived xenografts for personalized oncology. Asian J. Urol. 2016, 3, 240–253. [Google Scholar] [CrossRef]
- Pretlow, T.G.; Schwartz, S.; Giaconia, J.M.; Wright, A.L.; Grimm, H.A.; Edgehouse, N.L.; Murphy, J.R.; Markowitz, S.D.; Jamison, J.M.; Summers, J.L.; et al. Prostate cancer and other xenografts from cells in peripheral blood of patients. Cancer Res. 2000, 60, 4033–4036. [Google Scholar]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Kuske, A.; Joosse, S.A.; Yigit, G.; Sflomos, G.; Thaler, S.; Smit, D.J.; Werner, S.; Borgmann, K.; Gärtner, S.; et al. Characterization of circulating breast cancer cells with tumorigenic and metastatic capacity. EMBO Mol. Med. 2020, 12, e11908. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra148. [Google Scholar] [CrossRef]
- Zhao, P.; Zhou, W.; Liu, C.; Zhang, H.; Cheng, Z.; Wu, W.; Liu, K.; Hu, H.; Zhong, C.; Zhang, Y.; et al. Establishment and Characterization of a CTC Cell Line from Peripheral Blood of Breast Cancer Patient. J. Cancer 2019, 10, 6095–6104. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Park, S.H.; Jeon, C.H. Detection of circulating tumor cells in patients with breast cancer using the conditionally reprogrammed cell culture method and reverse transcription-PCR of hTERT and MAGE A1-6. Oncol. Lett. 2020, 20, 78. [Google Scholar] [CrossRef]
- Xiao, J.; McGill, J.R.; Stanton, K.; Kassner, J.D.; Choudhury, S.; Schlegel, R.; Sauna, Z.E.; Pohlmann, P.R.; Agarwal, S. Efficient Propagation of Circulating Tumor Cells: A First Step for Probing Tumor Metastasis. Cancers 2020, 12, 2784. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e114. [Google Scholar] [CrossRef]
- Wen, Y.; Mensah, N.N.; Song, X.; Zhu, J.; Tan, W.S.; Chen, X.; Li, J. A hydrogel with supramolecular surface functionalization for cancer cell capture and multicellular spheroid growth and release. Chem. Commun. 2022, 58, 681–684. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Z.; Ding, P.; Sun, N.; Liu, H.; Chen, Y.; Wang, Z.; Pei, R. Peptide NGR Modified TiO2 Nanofiber Substrate for Circulating Tumor Cells Capture. Adv. Fiber Mater. 2020, 2, 186–193. [Google Scholar] [CrossRef]
- Wu, L.; Ding, H.; Qu, X.; Shi, X.; Yang, J.; Huang, M.; Zhang, J.; Zhang, H.; Song, J.; Zhu, L.; et al. Fluidic Multivalent Membrane Nanointerface Enables Synergetic Enrichment of Circulating Tumor Cells with High Efficiency and Viability. J. Am. Chem. Soc. 2020, 142, 4800–4806. [Google Scholar] [CrossRef]
- Song, Y.; Shi, Y.; Huang, M.; Wang, W.; Wang, Y.; Cheng, J.; Lei, Z.; Zhu, Z.; Yang, C. Bioinspired Engineering of a Multivalent Aptamer-Functionalized Nanointerface to Enhance the Capture and Release of Circulating Tumor Cells. Angew. Chem. Int. Ed. Engl. 2019, 58, 2236–2240. [Google Scholar] [CrossRef] [PubMed]
- Cani, A.K.; Hayes, D.F. Breast Cancer Circulating Tumor Cells: Current Clinical Applications and Future Prospects. Clin. Chem. 2024, 70, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Riethdorf, S.; Müller, V.; Loibl, S.; Nekljudova, V.; Weber, K.; Huober, J.; Fehm, T.; Schrader, I.; Hilfrich, J.; Holms, F.; et al. Prognostic Impact of Circulating Tumor Cells for Breast Cancer Patients Treated in the Neoadjuvant “Geparquattro” Trial. Clin. Cancer Res. 2017, 23, 5384–5393. [Google Scholar] [CrossRef] [PubMed]
- Trapp, E.; Janni, W.; Schindlbeck, C.; Jückstock, J.; Andergassen, U.; de Gregorio, A.; Alunni-Fabbroni, M.; Tzschaschel, M.; Polasik, A.; Koch, J.G.; et al. Presence of Circulating Tumor Cells in High-Risk Early Breast Cancer During Follow-Up and Prognosis. J. Natl. Cancer Inst. 2019, 111, 380–387. [Google Scholar] [CrossRef]
- Thomas-Bonafos, T.; Pierga, J.Y.; Bidard, F.C.; Cabel, L.; Kiavue, N. Circulating tumor cells in breast cancer: Clinical validity and utility. npj Breast Cancer 2024, 10, 103. [Google Scholar] [CrossRef]
- Rack, B.; Schindlbeck, C.; Jückstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.; Lorenz, R.; Tesch, H.; Fasching, P.A.; et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014, 106, dju066. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Hayes, D.F.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Reuben, J.M.; Doyle, G.V.; Matera, J.; Allard, W.J.; Miller, M.C.; et al. Circulating tumor cells: A novel prognostic factor for newly diagnosed metastatic breast cancer. J. Clin. Oncol. 2005, 23, 1420–1430. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kiavue, N.; Jacot, W.; Bachelot, T.; Dureau, S.; Bourgeois, H.; Goncalves, A.; Brain, E.; Ladoire, S.; Dalenc, F.; et al. Overall Survival With Circulating Tumor Cell Count-Driven Choice of Therapy in Advanced Breast Cancer: A Randomized Trial. J. Clin. Oncol. 2024, 42, 383–389. [Google Scholar] [CrossRef]
- Nicolò, E.; Gianni, C.; Pontolillo, L.; Serafini, M.S.; Munoz-Arcos, L.S.; Andreopoulou, E.; Curigliano, G.; Reduzzi, C.; Cristofanilli, M. Circulating tumor cells et al.: Towards a comprehensive liquid biopsy approach in breast cancer. Transl. Breast Cancer Res. 2024, 5, 10. [Google Scholar] [CrossRef]
- Nicolò, E.; Serafini, M.S.; Munoz-Arcos, L.; Pontolillo, L.; Molteni, E.; Bayou, N.; Andreopoulou, E.; Curigliano, G.; Reduzzi, C.; Cristofanilli, M. Real-time assessment of HER2 status in circulating tumor cells of breast cancer patients: Methods of detection and clinical implications. J. Liq. Biopsy 2023, 2, 100117. [Google Scholar] [CrossRef]
- Pestrin, M.; Bessi, S.; Puglisi, F.; Minisini, A.M.; Masci, G.; Battelli, N.; Ravaioli, A.; Gianni, L.; Di Marsico, R.; Tondini, C.; et al. Final results of a multicenter phase II clinical trial evaluating the activity of single-agent lapatinib in patients with HER2-negative metastatic breast cancer and HER2-positive circulating tumor cells. A proof-of-concept study. Breast Cancer Res. Treat. 2012, 134, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Berger, F.; Cottu, P.; Loirat, D.; Rampanou, A.; Brain, E.; Cyrille, S.; Bourgeois, H.; Kiavue, N.; Deluche, E.; et al. Clinical utility of circulating tumour cell-based monitoring of late-line chemotherapy for metastatic breast cancer: The randomised CirCe01 trial. Br. J. Cancer 2021, 124, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Fehm, T.; Mueller, V.; Banys-Paluchowski, M.; Fasching, P.A.; Friedl, T.W.P.; Hartkopf, A.; Huober, J.; Loehberg, C.; Rack, B.; Riethdorf, S.; et al. Efficacy of Lapatinib in Patients with HER2-Negative Metastatic Breast Cancer and HER2-Positive Circulating Tumor Cells-The DETECT III Clinical Trial. Clin. Chem. 2024, 70, 307–318. [Google Scholar] [CrossRef]
- Smit, D.J.; Pantel, K.; Jücker, M. Circulating tumor cells as a promising target for individualized drug susceptibility tests in cancer therapy. Biochem. Pharmacol. 2021, 188, 114589. [Google Scholar] [CrossRef]
- Mu, H.Y.; Ou, Y.C.; Chuang, H.N.; Lu, T.J.; Jhan, P.P.; Hsiao, T.H.; Huang, J.H. Triple Selection Strategy for In Situ Labeling of Circulating Tumor Cells with High Purity and Viability toward Preclinical Personalized Drug Sensitivity Analysis. Adv. Biosyst. 2020, 4, e2000013. [Google Scholar] [CrossRef]
- Gerratana, L.; Davis, A.A.; Zhang, Q.; Basile, D.; Rossi, G.; Strickland, K.; Franzoni, A.; Allegri, L.; Mu, Z.; Zhang, Y.; et al. Longitudinal Dynamics of Circulating Tumor Cells and Circulating Tumor DNA for Treatment Monitoring in Metastatic Breast Cancer. JCO Precis. Oncol. 2021, 5, 943–952. [Google Scholar] [CrossRef]
- Liu, M.C.; MacKay, M.; Kase, M.; Piwowarczyk, A.; Lo, C.; Schaeffer, J.; Finkle, J.D.; Mason, C.E.; Beaubier, N.; Blackwell, K.L.; et al. Longitudinal Shifts of Solid Tumor and Liquid Biopsy Sequencing Concordance in Metastatic Breast Cancer. JCO Precis. Oncol. 2022, 6, e2100321. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Carneiro, B.A.; Chandra, S.; Mohindra, N.; Kalyan, A.; Kaplan, J.; Matsangou, M.; Pai, S.; Costa, R.; et al. Concordance between genomic alterations assessed by next-generation sequencing in tumor tissue or circulating cell-free DNA. Oncotarget 2016, 7, 65364–65373. [Google Scholar] [CrossRef]
- Finkle, J.D.; Boulos, H.; Driessen, T.M.; Lo, C.; Blidner, R.A.; Hafez, A.; Khan, A.A.; Lozac’hmeur, A.; McKinnon, K.E.; Perera, J.; et al. Validation of a liquid biopsy assay with molecular and clinical profiling of circulating tumor DNA. npj Precis. Oncol. 2021, 5, 63. [Google Scholar] [CrossRef]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Patel, S.P.; Harismendy, O.; Ikeda, M.; Parker, B.A.; et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clin. Cancer Res. 2016, 22, 5497–5505. [Google Scholar] [CrossRef] [PubMed]
- Zundelevich, A.; Dadiani, M.; Kahana-Edwin, S.; Itay, A.; Sella, T.; Gadot, M.; Cesarkas, K.; Farage-Barhom, S.; Saar, E.G.; Eyal, E.; et al. ESR1 mutations are frequent in newly diagnosed metastatic and loco-regional recurrence of endocrine-treated breast cancer and carry worse prognosis. Breast Cancer Res. 2020, 22, 16. [Google Scholar] [CrossRef] [PubMed]
- Campanile, F.; Maurea, S.; Mainenti, P.; Corvino, A.; Imbriaco, M. Duodenal Involvement by Breast Cancer. Breast J. 2012, 18, 615–616. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e456. [Google Scholar] [CrossRef]
- Janda, C.Y.; Dang, L.T.; You, C.; Chang, J.; de Lau, W.; Zhong, Z.A.; Yan, K.S.; Marecic, O.; Siepe, D.; Li, X.; et al. Surrogate Wnt agonists that phenocopy canonical Wnt and β-catenin signalling. Nature 2017, 545, 234–237. [Google Scholar] [CrossRef]
- Mihara, E.; Hirai, H.; Yamamoto, H.; Tamura-Kawakami, K.; Matano, M.; Kikuchi, A.; Sato, T.; Takagi, J. Active and water-soluble form of lipidated Wnt protein is maintained by a serum glycoprotein afamin/α-albumin. eLife 2016, 5, e11621. [Google Scholar] [CrossRef]
- Artegiani, B.; Hendriks, D.; Beumer, J.; Kok, R.; Zheng, X.; Joore, I.; Chuva de Sousa Lopes, S.; van Zon, J.; Tans, S.; Clevers, H. Fast and efficient generation of knock-in human organoids using homology-independent CRISPR-Cas9 precision genome editing. Nat. Cell Biol. 2020, 22, 321–331. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Amatngalim, G.D.; Oka, R.; Meijers, F.M.; Kruisselbrink, E.; van Mourik, P.; Berkers, G.; de Winter-de Groot, K.M.; Michel, S.; et al. CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 2020, 26, 503–510.e507. [Google Scholar] [CrossRef]
- Michels, B.E.; Mosa, M.H.; Streibl, B.I.; Zhan, T.; Menche, C.; Abou-El-Ardat, K.; Darvishi, T.; Członka, E.; Wagner, S.; Winter, J.; et al. Pooled In Vitro and In Vivo CRISPR-Cas9 Screening Identifies Tumor Suppressors in Human Colon Organoids. Cell Stem Cell 2020, 26, 782–792.e787. [Google Scholar] [CrossRef]
- Ringel, T.; Frey, N.; Ringnalda, F.; Janjuha, S.; Cherkaoui, S.; Butz, S.; Srivatsa, S.; Pirkl, M.; Russo, G.; Villiger, L.; et al. Genome-Scale CRISPR Screening in Human Intestinal Organoids Identifies Drivers of TGF-β Resistance. Cell Stem Cell 2020, 26, 431–440.e438. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).