
Integrative Analysis of DNA Methylation and microRNA Reveals GNPDA1 and SLC25A16 Related to Biopsychosocial Factors Among Taiwanese Women with a Family History of Breast Cancer

 ,
,  ,
,  , ,
, ,  ,
,  ,
,
Abstract
1. Introduction
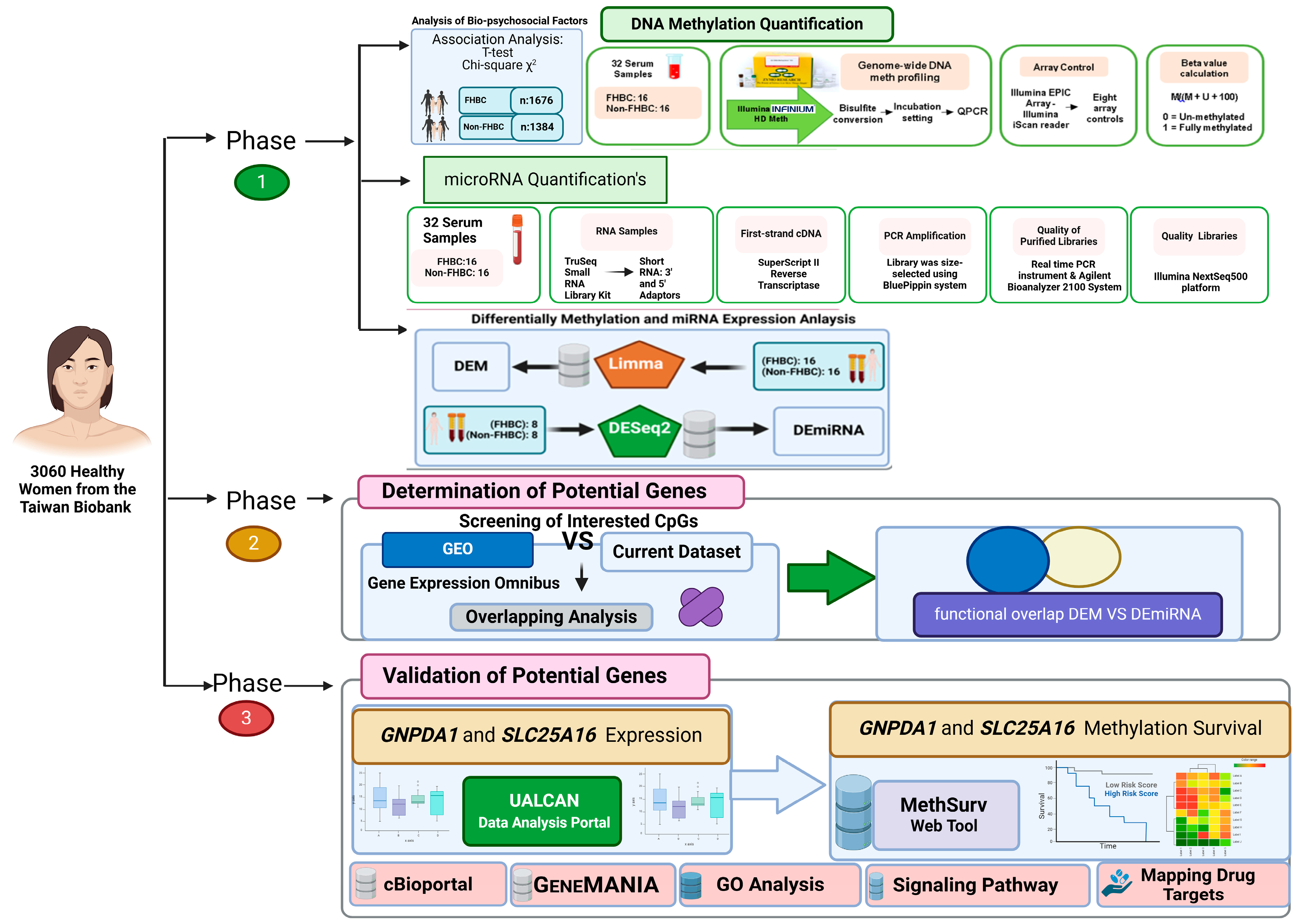
2. Materials and Methods
2.1. Study Design
2.2. Study Population
2.3. Analysis of Biopsychosocial Factors
2.4. DNA Methylation and miRNA Quantification
2.5. Differential Analysis of Methylated DNA and miRNA
2.6. Determination of Potential Genes
2.6.1. Phase One: Functional Overlap Between DEM Genes in the Current Study and GEO Database
2.6.2. Phase Two: Functional Overlap Between Phase One’s DEM Genes and DEmiRNA to Reveal Potential Genes
2.7. Prognostic Value of Potential Genes in Breast Cancer
2.7.1. Gene Expression Profiles: UALCAN Analysis
2.7.2. Survival Analysis of Two Potential Genes
2.7.3. cBioPortal and GeneMANIA Analysis of Two Potential Genes
2.7.4. Functional Enrichment Analysis of Potential Genes
2.7.5. Mapping Drug Targets for Breast Cancer Using Drug Database
3. Results
3.1. Population Demographics
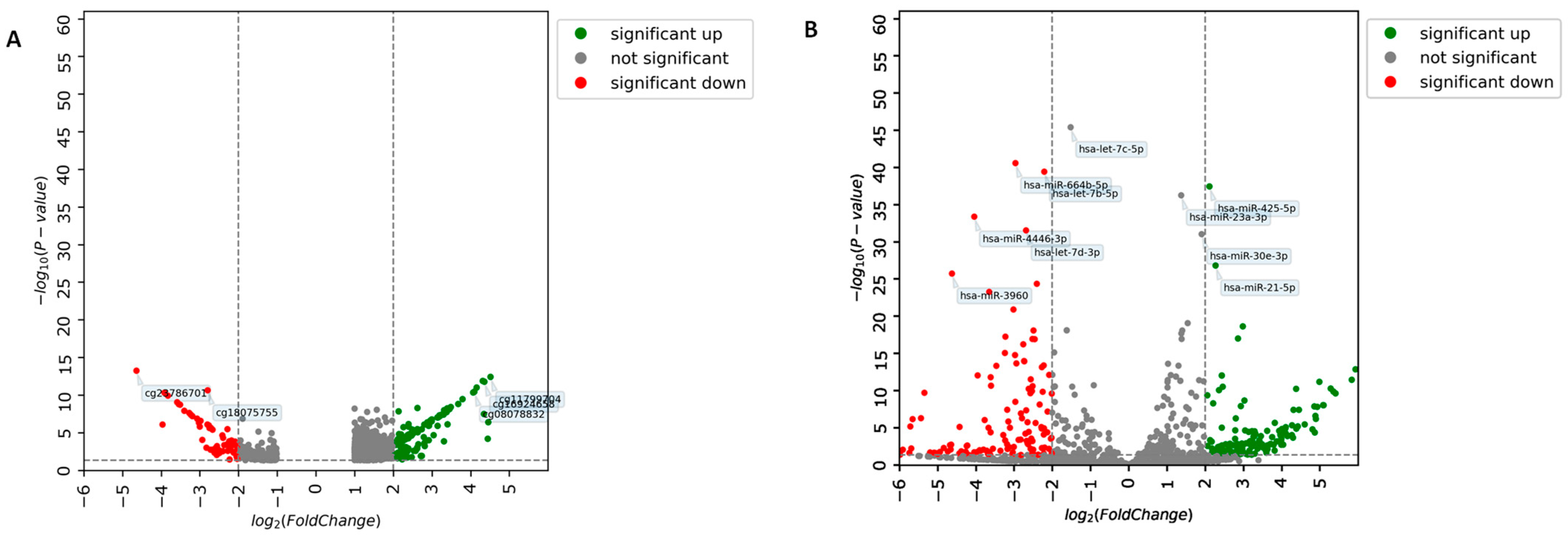
3.2. Identification of DEM and DEmiRNA
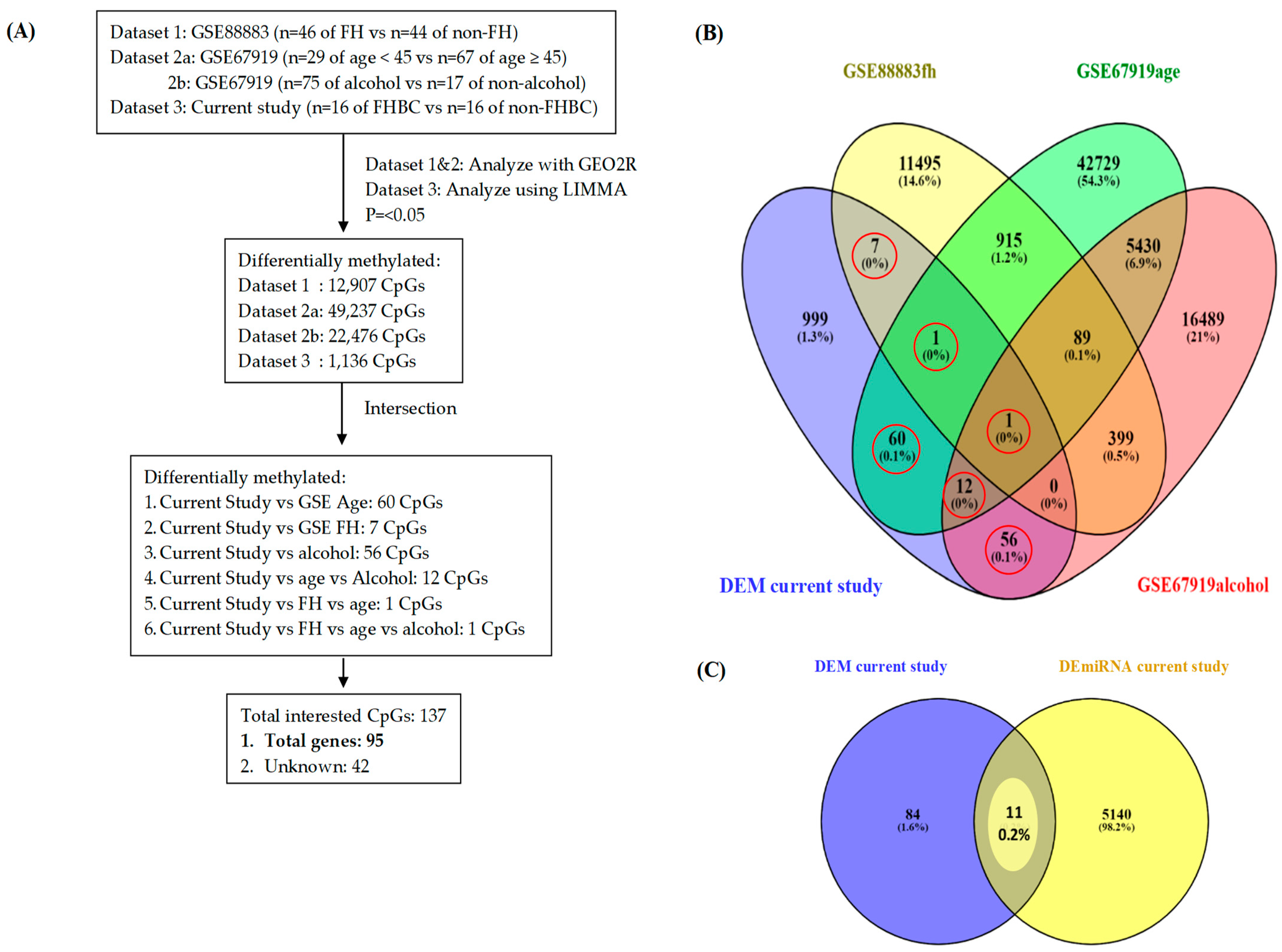
3.3. Identification of Potential Genes (GEO Database and Current Study)
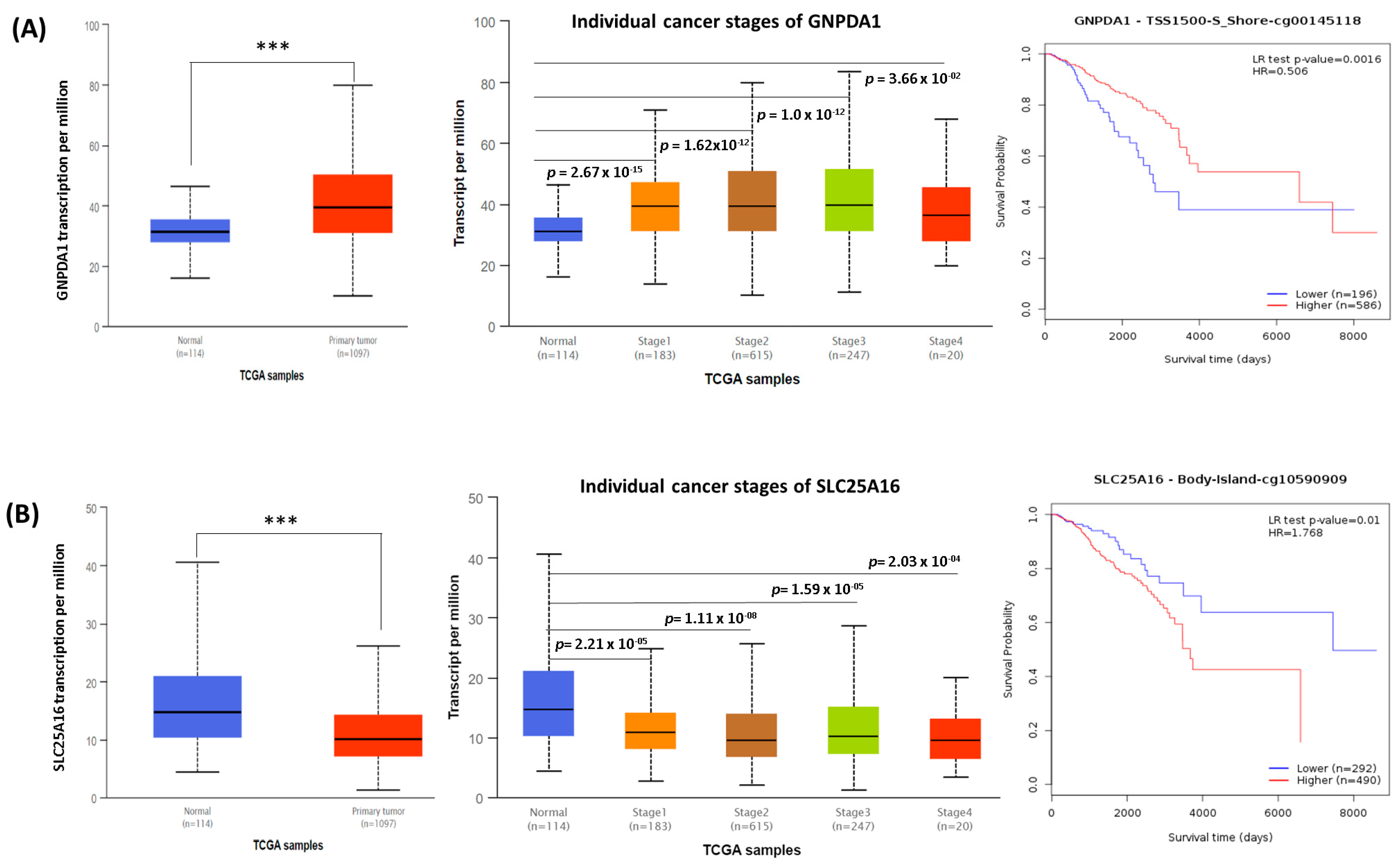
3.4. The Role of Potential Genes in Breast Cancer Prognosis (TCGA Database)
3.5. Genetic Alteration, Gene Interaction, and Neighbor Gene Network of Two Potential Genes in Patients with Breast Cancer
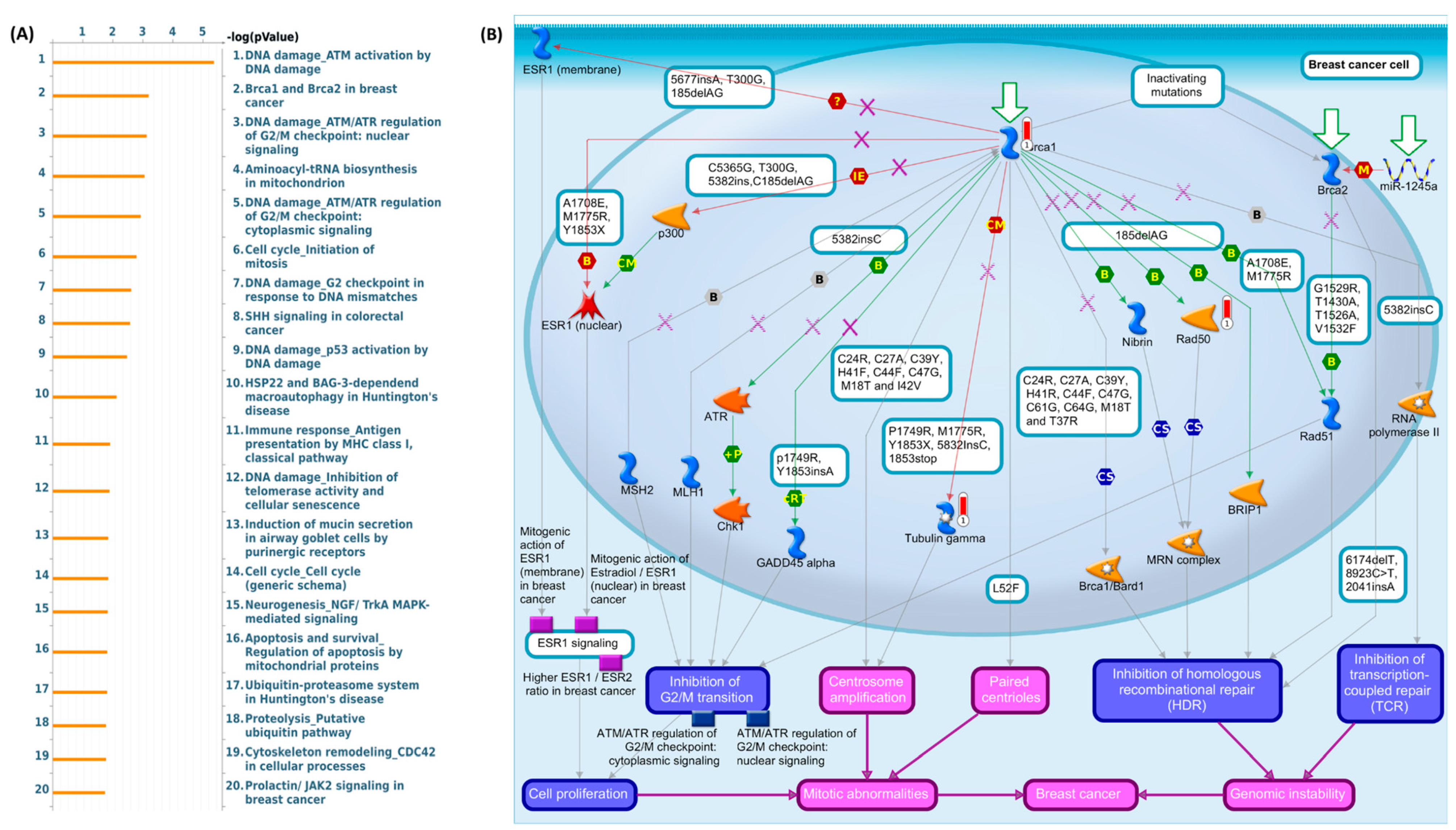
3.6. Gene Ontology, KEGG, and Genome Pathway Exploration of Two Potential Genes in Breast Cancer
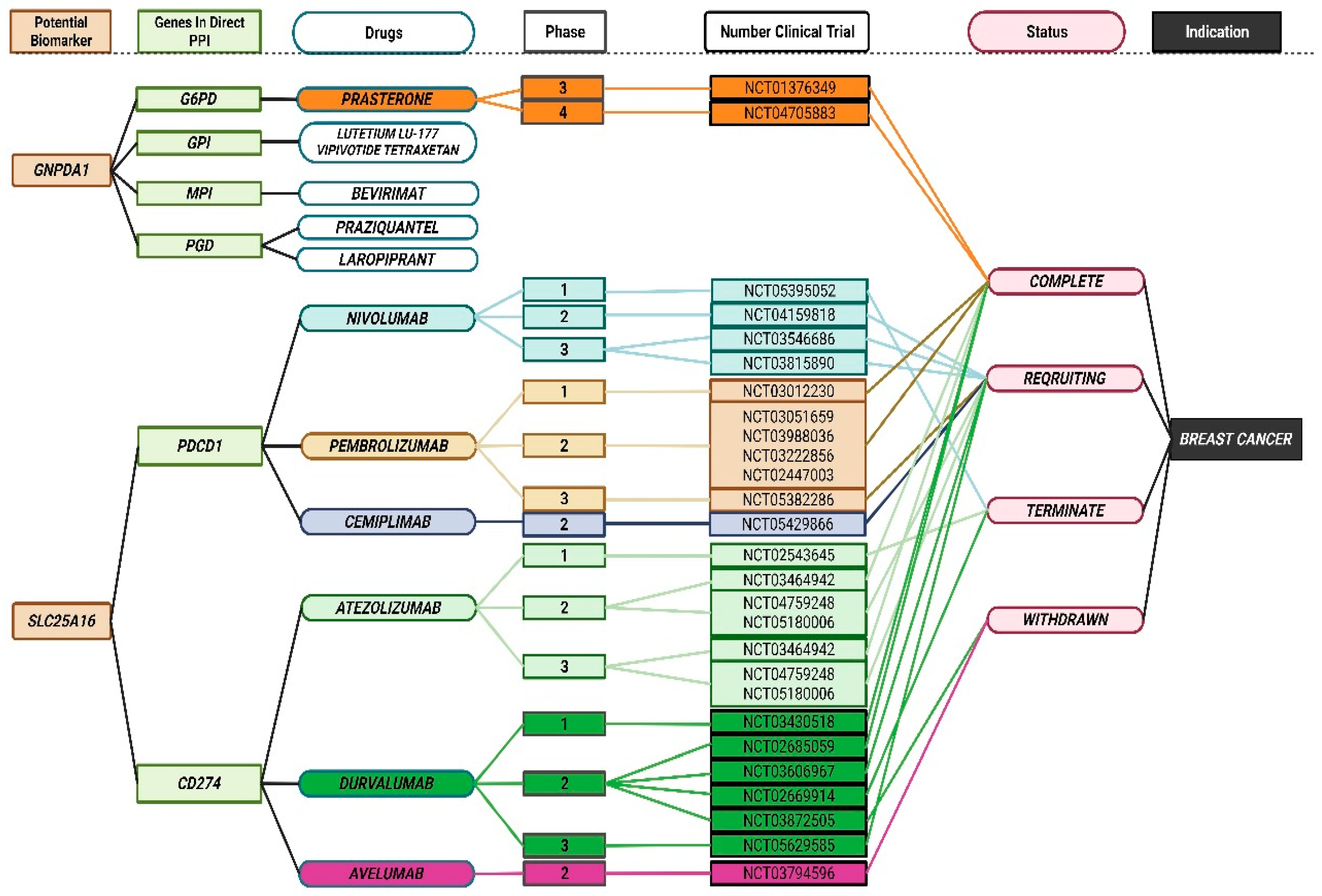
3.7. Discovery of Breast Cancer Drug Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FHBC | Family History Of Breast Cancer |
| miRNA | microRNA |
| DEM | Differentially Methylated |
| DEmiRNA | Differential Expressed microRNA |
| LIMMA | Linear Models For Microarray |
| DESeq2 | Differential Gene Expression Analysis Of RNA-Seq 2 |
| GEO | Gene Expression Omnibus |
| mRNA | Messenger RNA |
| cBioPortal | cBio Cancer Genomics Portal |
| KEGG | Kyoto Encyclopedia Of Genes And Genomes |
| PPIs | Protein–Protein Interactions |
| BMI | Body Mass Index |
| FDR | False Discovery Rate |
| BH | Benjamini–Hochberg |
References
- Johnson, K.C.; Houseman, E.A.; King, J.E.; Christensen, B.C. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. 2017, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.J.; Han, W. A Review of the Epidemiology of Breast Cancer in Asia: Focus on Risk Factors. Asian Pac. J. Cancer Prev. 2020, 21, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Winters, S.; Martin, C.; Murphy, D.; Shokar, N.K. Breast Cancer Epidemiology, Prevention, and Screening. Prog. Mol. Biol. Transl. Sci. 2017, 151, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.S.; Zhao, Z.; Yang, Z.N.; Xu, F.; Lu, H.J.; Zhu, Z.Y.; Shi, W.; Jiang, J.; Yao, P.P.; Zhu, H.P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef]
- Holm, J.; Eriksson, L.; Ploner, A.; Eriksson, M.; Rantalainen, M.; Li, J.; Hall, P.; Czene, K. Assessment of Breast Cancer Risk Factors Reveals Subtype Heterogeneity. Cancer Res. 2017, 77, 3708–3717. [Google Scholar] [CrossRef]
- Zendehbad, Z.; Izadi, P.; Daraei, A.; Yekaninejad, M.S.; Nafissi, N.; Younosi, N.; Khorasani, G.; Tavakkoly Bazzaz, J. Early Parity Epigenetic Footprint of FOXA1 Gene Body in Normal Breast Tissue of Iranian Women. Iran. Biomed. J. 2019, 23, 99–106. [Google Scholar] [CrossRef]
- Engmann, N.J.; Golmakani, M.K.; Miglioretti, D.L.; Sprague, B.L.; Kerlikowske, K. Population-Attributable Risk Proportion of Clinical Risk Factors for Breast Cancer. JAMA Oncol. 2017, 3, 1228–1236. [Google Scholar] [CrossRef]
- Wu, H.C.; Do, C.; Andrulis, I.L.; John, E.M.; Daly, M.B.; Buys, S.S.; Chung, W.K.; Knight, J.A.; Bradbury, A.R.; Keegan, T.H.M.; et al. Breast cancer family history and allele-specific DNA methylation in the legacy girls study. Epigenetics 2018, 13, 240–250. [Google Scholar] [CrossRef]
- Chen, M.; Wong, E.M.; Nguyen, T.L.; Dite, G.S.; Stone, J.; Dugué, P.A.; Giles, G.G.; Southey, M.C.; Milne, R.L.; Hopper, J.L.; et al. DNA methylation-based biological age, genome-wide average DNA methylation, and conventional breast cancer risk factors. Sci. Rep. 2019, 9, 15055. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, M.; Ke, Z.J.; Luo, J. Cellular and molecular mechanisms underlying alcohol-induced aggressiveness of breast cancer. Pharmacol. Res. 2017, 115, 299–308. [Google Scholar] [CrossRef]
- Conway, K.; Edmiston, S.N.; Parrish, E.; Bryant, C.; Tse, C.K.; Swift-Scanlan, T.; McCullough, L.E.; Kuan, P.F. Breast tumor DNA methylation patterns associated with smoking in the Carolina Breast Cancer Study. Breast Cancer Res. Treat. 2017, 163, 349–361. [Google Scholar] [CrossRef]
- Varghese, R.S.; Barefoot, M.E.; Jain, S.; Chen, Y.; Zhang, Y.; Alley, A.; Kroemer, A.H.; Tadesse, M.G.; Kumar, D.; Sherif, Z.A.; et al. Integrative Analysis of DNA Methylation and microRNA Expression Reveals Mechanisms of Racial Heterogeneity in Hepatocellular Carcinoma. Front. Genet. 2021, 12, 708326. [Google Scholar] [CrossRef]
- Lehmann, U. Aberrant DNA methylation of microRNA genes in human breast cancer—A critical appraisal. Cell Tissue Res. 2014, 356, 657–664. [Google Scholar] [CrossRef]
- Aure, M.R.; Fleischer, T.; Bjørklund, S.; Ankill, J.; Castro-Mondragon, J.A.; Børresen-Dale, A.L.; Tost, J.; Sahlberg, K.K.; Mathelier, A.; Tekpli, X.; et al. Crosstalk between microRNA expression and DNA methylation drives the hormone-dependent phenotype of breast cancer. Genome Med. 2021, 13, 72. [Google Scholar] [CrossRef]
- Feng, Y.-C.A.; Chen, C.-Y.; Chen, T.-T.; Kuo, P.-H.; Hsu, Y.-H.; Yang, H.-I.; Chen, W.J.; Shen, C.-Y.; Ge, T.; Huang, H.; et al. Taiwan Biobank: A rich biomedical research database of the Taiwanese population. medRxiv 2021, 2, 100197. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Dong, X.; Tian, L.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 2016, 5, ISCB. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Li, Y.; Gu, J.; Xu, F.; Zhu, Q.; Ge, D.; Lu, C. Transcriptomic and functional network features of lung squamous cell carcinoma through integrative analysis of GEO and TCGA data. Sci. Rep. 2018, 8, 15834. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Modhukur, V.; Iljasenko, T.; Metsalu, T.; Lokk, K.; Laisk-Podar, T.; Vilo, J. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 2018, 10, 277–288. [Google Scholar] [CrossRef]
- Wu, P.; Heins, Z.J.; Muller, J.T.; Katsnelson, L.; de Bruijn, I.; Abeshouse, A.A.; Schultz, N.; Fenyö, D.; Gao, J. Integration and Analysis of CPTAC Proteomics Data in the Context of Cancer Genomics in the cBioPortal. Mol. Cell Proteom. 2019, 18, 1893–1898. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.H.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y.D. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS ONE 2017, 12, e0184129. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Tas, F.; Keskin, S. Age-specific incidence ratios of breast cancer (BC) in Turkey: BC in older people is increasing. Arch. Gerontol. Geriatr. 2012, 55, 112–115. [Google Scholar] [CrossRef]
- Barginear, M.F.; Muss, H.; Kimmick, G.; Owusu, C.; Mrozek, E.; Shahrokni, A.; Ballman, K.; Hurria, A. Breast cancer and aging: Results of the U13 conference breast cancer panel. Breast Cancer Res. Treat. 2014, 146, 1–6. [Google Scholar] [CrossRef]
- Rojas, K.; Stuckey, A. Breast Cancer Epidemiology and Risk Factors. Clin. Obstet. Gynecol. 2016, 59, 651–672. [Google Scholar] [CrossRef]
- Ellingjord-Dale, M.; Vos, L.; Tretli, S.; Hofvind, S.; Dos-Santos-Silva, I.; Ursin, G. Parity, hormones and breast cancer subtypes—Results from a large nested case-control study in a national screening program. Breast Cancer Res. 2017, 19, 10. [Google Scholar] [CrossRef] [PubMed]
- Özkan, M.; Yıldırım, N.; Dişçi, R.; İlgün, A.S.; Sarsenov, D.; Alço, G.; Aktepe, F.; Kalyoncu, N.; İzci, F.; Selamoğlu, D.; et al. Roles of Biopsychosocial Factors in the Development of Breast Cancer. Eur. J. Breast Health 2017, 13, 206–212. [Google Scholar] [CrossRef]
- McPherson, K.; Steel, C.M.; Dixon, J.M. ABC of breast diseases. Breast cancer--epidemiology, risk factors and genetics. BMJ 1994, 309, 1003–1006. [Google Scholar] [CrossRef]
- Freudenheim, J.L. Alcohol’s Effects on Breast Cancer in Women. Alcohol. Res. 2020, 40, 11. [Google Scholar] [CrossRef]
- Carreras, G.; Lachi, A.; Boffi, R.; Clancy, L.; Gallus, S.; Fernández, E.; López, M.J.; Soriano, J.B.; López Nicolás, Á.; Semple, S.; et al. Burden of disease from breast cancer attributable to smoking and second-hand smoke exposure in Europe. Int. J. Cancer 2020, 147, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Vona-Davis, L.; Rose, D.P. The influence of socioeconomic disparities on breast cancer tumor biology and prognosis: A review. J. Womens Health 2009, 18, 883–893. [Google Scholar] [CrossRef]
- Dailey, A.B.; Kasl, S.V.; Holford, T.R.; Calvocoressi, L.; Jones, B.A. Neighborhood-level socioeconomic predictors of nonadherence to mammography screening guidelines. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2293–2303. [Google Scholar] [CrossRef]
- Li, D.; Cheng, X.; Zheng, W.; Chen, J. Glucosamine-6-Phosphate Isomerase 1 Promotes Tumor Progression and Indicates Poor Prognosis in Hepatocellular Carcinoma. Cancer Manag. Res. 2020, 12, 4923–4935. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Zhu, Y.; Qin, S. Glucose-6-phosphate dehydrogenase: A biomarker and potential therapeutic target for cancer. Anticancer. Agents Med. Chem. 2014, 14, 280–289. [Google Scholar] [CrossRef]
- Chen, L.Y.; Huang, R.L.; Su, P.H.; Chu, L.H.; Weng, Y.C.; Wang, H.C.; Lai, H.C.; Wen, K.C. Epigenomic Profiling of Epithelial Ovarian Cancer Stem-Cell Differentiation Reveals GPD1 Associated Immune Suppressive Microenvironment and Poor Prognosis. Int. J. Mol. Sci. 2022, 23, 5120. [Google Scholar] [CrossRef]
- Xia, R.; Tang, H.; Shen, J.; Xu, S.; Liang, Y.; Zhang, Y.; Gong, X.; Min, Y.; Zhang, D.; Tao, C.; et al. Prognostic value of a novel glycolysis-related gene expression signature for gastrointestinal cancer in the Asian population. Cancer Cell Int. 2021, 21, 154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, J.; Wang, S.; Jiang, Q.; Xu, K. Identification and Validation of a Nine-Gene Amino Acid Metabolism-Related Risk Signature in HCC. Front. Cell Dev. Biol. 2021, 9, 731790. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, S.; Cai, Z.; Gao, F.; Deng, W.; Wen, Y.; Qiu, Z.W.; Hou, Z.K.; Chen, X.L. A glycolysis-related gene pairs signature predicts prognosis in patients with hepatocellular carcinoma. PeerJ 2020, 8, e9944. [Google Scholar] [CrossRef]
- Saviana, M.; Le, P.; Micalo, L.; Del Valle-Morales, D.; Romano, G.; Acunzo, M.; Li, H.; Nana-Sinkam, P. Crosstalk between miRNAs and DNA Methylation in Cancer. Genes 2023, 14, 1075. [Google Scholar] [CrossRef]
- Khan, S.; Ansar, M.; Khan, A.K.; Shah, K.; Muhammad, N.; Shahzad, S.; Nickerson, D.A.; Bamshad, M.J.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. A homozygous missense mutation in SLC25A16 associated with autosomal recessive isolated fingernail dysplasia in a Pakistani family. Br. J. Dermatol. 2018, 178, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, R.G.; Marian, C.; Krishnan, S.S.; Spear, S.L.; Kallakury, B.V.; Perry, D.J.; Convit, J.R.; Seillier-Moiseiwitsch, F.; Yang, Y.; Freudenheim, J.L.; et al. Familial and racial determinants of tumour suppressor genes promoter hypermethylation in breast tissues from healthy women. J. Cell Mol. Med. 2010, 14, 1468–1475. [Google Scholar] [CrossRef]
- Wu, H.C.; Southey, M.C.; Hibshoosh, H.; Santella, R.M.; Terry, M.B. DNA Methylation in Breast Tumor from High-risk Women in the Breast Cancer Family Registry. Anticancer Res. 2017, 37, 659–664. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, L.; Wang, L.; Xiao, J.; Sun, J.; Zhou, Y.; Xu, X.; Xu, W.; Spiliopoulou, A.; Timofeeva, M.; et al. Alcohol consumption, blood DNA methylation and breast cancer: A Mendelian randomisation study. Eur. J. Epidemiol. 2022, 37, 701–712. [Google Scholar] [CrossRef]
- Farina, N.H.; Ramsey, J.E.; Cuke, M.E.; Ahern, T.P.; Shirley, D.J.; Stein, J.L.; Stein, G.S.; Lian, J.B.; Wood, M.E. Development of a predictive miRNA signature for breast cancer risk among high-risk women. Oncotarget 2017, 8, 112170–112183. [Google Scholar] [CrossRef]
- Bao, L.; Zhao, J.; Dai, X.; Wang, Y.; Ma, R.; Su, Y.; Cui, H.; Niu, J.; Bai, S.; Xiao, Z.; et al. Correlation between miR-23a and onset of hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Ali-Eldin, Z.A.; Elbedewy, T.A.; El-Serafy, M.; Ali-Eldin, F.A.; AbdelAziz, H. MicroRNAs and clinical implications in hepatocellular carcinoma. World J. Hepatol. 2017, 9, 1001–1007. [Google Scholar] [CrossRef]
- Todeschini, P.; Salviato, E.; Romani, C.; Raimondi, V.; Ciccarese, F.; Ferrari, F.; Tognon, G.; Marchini, S.; D’Incalci, M.; Zanotti, L.; et al. Comprehensive Profiling of Hypoxia-Related miRNAs Identifies miR-23a-3p Overexpression as a Marker of Platinum Resistance and Poor Prognosis in High-Grade Serous Ovarian Cancer. Cancers 2021, 13, 3358. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, X.; Xu, W.; Zhou, P.; Gao, P.; Jiang, S.; Lobie, P.E.; Zhu, T. c-MYC-regulated miR-23a/24-2/27a cluster promotes mammary carcinoma cell invasion and hepatic metastasis by targeting Sprouty2. J. Biol. Chem. 2013, 288, 18121–18133. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Zhang, H.; Kim, J.; Getz, G.; Whetstine, J.R. Regulation of Transient Site-specific Copy Gain by MicroRNA. J. Biol. Chem. 2016, 291, 4862–4871. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, J.; Jiang, H.; Dong, H. A Novel Necroptosis-Related miRNA Signature for Predicting the Prognosis of Breast Cancer Metastasis. Dis. Markers 2022, 2022, 3391878. [Google Scholar] [CrossRef]
- Gu, C.; Hou, C.; Zhang, S. miR-425-5p improves inflammation and septic liver damage through negatively regulating the RIP1-mediated necroptosis. Inflamm. Res. 2020, 69, 299–308. [Google Scholar] [CrossRef]
- Lee, E.Y.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef]
- Martín-Lorenzo, A.; Gonzalez-Herrero, I.; Rodríguez-Hernández, G.; García-Ramírez, I.; Vicente-Dueñas, C.; Sánchez-García, I. Early epigenetic cancer decisions. Biol. Chem. 2014, 395, 1315–1320. [Google Scholar] [CrossRef]
- Liu, Y.; Colditz, G.A.; Gehlert, S.; Goodman, M. Racial disparities in risk of second breast tumors after ductal carcinoma in situ. Breast Cancer Res. Treat. 2014, 148, 163–173. [Google Scholar] [CrossRef]
- Dania, V.; Liu, Y.; Ademuyiwa, F.; Weber, J.D.; Colditz, G.A. Associations of race and ethnicity with risk of developing invasive breast cancer after lobular carcinoma in situ. Breast Cancer Res. 2019, 21, 120. [Google Scholar] [CrossRef]
- Sweeney, C.; Bernard, P.S.; Factor, R.E.; Kwan, M.L.; Habel, L.A.; Quesenberry, C.P., Jr.; Shakespear, K.; Weltzien, E.K.; Stijleman, I.J.; Davis, C.A.; et al. Intrinsic subtypes from PAM50 gene expression assay in a population-based breast cancer cohort: Differences by age, race, and tumor characteristics. Cancer Epidemiol. Biomark. Prev. 2014, 23, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, M.; Lin, Y.; Zhao, J.; Gu, H.; Li, X. Circulating tumor DNA methylation: A promising clinical tool for cancer diagnosis and management. Clin. Chem. Lab. Med. 2024, 62, 2111–2127. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J. Natl. Cancer Inst. 2017, 109, djx118. [Google Scholar] [CrossRef] [PubMed]
- Gahlawat, A.W.; Witte, T.; Sinn, P.; Schott, S. Circulating cf-miRNA as a more appropriate surrogate liquid biopsy marker than cfDNA for ovarian cancer. Sci. Rep. 2023, 13, 5503. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Li, W.L.; Liu, B.H.; Dong, H.; Mou, Z.R.; Wu, Y.Z. Identification of differential proteins in colorectal cancer cells treated with caffeic acid phenethyl ester. World J. Gastroenterol. 2014, 20, 11840–11849. [Google Scholar] [CrossRef]
- Wang, X.; Cimermancic, P.; Yu, C.; Schweitzer, A.; Chopra, N.; Engel, J.L.; Greenberg, C.; Huszagh, A.S.; Beck, F.; Sakata, E.; et al. Molecular Details Underlying Dynamic Structures and Regulation of the Human 26S Proteasome. Mol. Cell Proteom. 2017, 16, 840–854. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Agten, S.M.; Eckardt, V.; Blanchet, X.; Schmitt, M.M.; Ippel, H.; Neideck, C.; Bidzhekov, K.; Leberzammer, J.; Wichapong, K.; et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 2017, 9, eaah6650. [Google Scholar] [CrossRef]
- McFarland, M.A.; Ellis, C.E.; Markey, S.P.; Nussbaum, R.L. Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol. Cell Proteom. 2008, 7, 2123–2137. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Liu, Y.; Lauritzen, K.H.; Erdjument-Bromage, H.; Martin, B.; Swayne, T.C.; Jiang, X.; Przedborski, S. PINK1 Content in Mitochondria is Regulated by ER-Associated Degradation. J. Neurosci. 2019, 39, 7074–7085. [Google Scholar] [CrossRef]
- Rubio, A.J.; Bencomo-Alvarez, A.E.; Young, J.E.; Velazquez, V.V.; Lara, J.J.; Gonzalez, M.A.; Eiring, A.M. 26S Proteasome Non-ATPase Regulatory Subunits 1 (PSMD1) and 3 (PSMD3) as Putative Targets for Cancer Prognosis and Therapy. Cells 2021, 10, 2390. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Kim, E.S.; Koo, J.S. Amino Acid Transporters and Glutamine Metabolism in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 907. [Google Scholar] [CrossRef]
- Terao, M.; Goracci, L.; Celestini, V.; Kurosaki, M.; Bolis, M.; Di Veroli, A.; Vallerga, A.; Fratelli, M.; Lupi, M.; Corbelli, A.; et al. Role of mitochondria and cardiolipins in growth inhibition of breast cancer cells by retinoic acid. J. Exp. Clin. Cancer Res. 2019, 38, 436. [Google Scholar] [CrossRef]
- Iino, K.; Mitobe, Y.; Ikeda, K.; Takayama, K.I.; Suzuki, T.; Kawabata, H.; Suzuki, Y.; Horie-Inoue, K.; Inoue, S. RNA-binding protein NONO promotes breast cancer proliferation by post-transcriptional regulation of SKP2 and E2F8. Cancer Sci. 2020, 111, 148–159. [Google Scholar] [CrossRef]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liang, Y.L.; Liu, Y.; Chen, Y.Y.; Yang, S.T.; Li, B.R.; Yu, Y.X.; Lyu, Y.; Wang, R. MeCP2 inhibits proliferation and migration of breast cancer via suppression of epithelial-mesenchymal transition. J. Cell Mol. Med. 2020, 24, 7959–7967. [Google Scholar] [CrossRef]
- Hlaváč, V.; Václavíková, R.; Brynychová, V.; Koževnikovová, R.; Kopečková, K.; Vrána, D.; Gatěk, J.; Souček, P. Role of Genetic Variation in ABC Transporters in Breast Cancer Prognosis and Therapy Response. Int. J. Mol. Sci. 2020, 21, 9556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Deng, H.; Wang, Z.Y. Estrogen activation of the mitogen-activated protein kinase is mediated by ER-α36 in ER-positive breast cancer cells. J. Steroid Biochem. Mol. Biol. 2014, 143, 434–443. [Google Scholar] [CrossRef]
- Rizza, P.; Barone, I.; Zito, D.; Giordano, F.; Lanzino, M.; De Amicis, F.; Mauro, L.; Sisci, D.; Catalano, S.; Dahlman Wright, K.; et al. Estrogen receptor beta as a novel target of androgen receptor action in breast cancer cell lines. Breast Cancer Res. 2014, 16, R21. [Google Scholar] [CrossRef]
- He, J.; Chen, Y.; Cai, L.; Li, Z.; Guo, X. UBAP2L silencing inhibits cell proliferation and G2/M phase transition in breast cancer. Breast Cancer 2018, 25, 224–232. [Google Scholar] [CrossRef]
- Golrokh Mofrad, M.; Sadigh, Z.A.; Ainechi, S.; Faghihloo, E. Detection of human papillomavirus genotypes, herpes simplex, varicella zoster and cytomegalovirus in breast cancer patients. Virol. J. 2021, 18, 25. [Google Scholar] [CrossRef]
- Teshigahara, O.; Goshima, F.; Takao, K.; Kohno, S.; Kimata, H.; Nakao, A.; Nishiyama, Y. Oncolytic viral therapy for breast cancer with herpes simplex virus type 1 mutant HF 10. J. Surg. Oncol. 2004, 85, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.; Vijay, G.V.; Soundararajan, R.; Yu, X.; Symmans, W.F.; Sphyris, N.; Mani, S.A. FOXC2 regulates the G2/M transition of stem cell-rich breast cancer cells and sensitizes them to PLK1 inhibition. Sci. Rep. 2016, 6, 23070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, J.; Qu, C.; Peng, Y.; Lei, J.; Sun, L.; Zong, B.; Liu, S. A look into the link between centrosome amplification and breast cancer. Biomed. Pharmacother. 2020, 132, 110924. [Google Scholar] [CrossRef]
- Piemonte, K.M.; Anstine, L.J.; Keri, R.A. Centrosome Aberrations as Drivers of Chromosomal Instability in Breast Cancer. Endocrinology 2021, 162, bqab208. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, J.L.; D’Assoro, A.B.; Lingle, W.L. Centrosome amplification and the origin of chromosomal instability in breast cancer. J. Mammary Gland. Biol. Neoplasia 2004, 9, 275–283. [Google Scholar] [CrossRef]
- Singh, A.; Denu, R.A.; Wolfe, S.K.; Sperger, J.M.; Schehr, J.; Witkowsky, T.; Esbona, K.; Chappell, R.J.; Weaver, B.A.; Burkard, M.E.; et al. Centrosome amplification is a frequent event in circulating tumor cells from subjects with metastatic breast cancer. Mol. Oncol. 2020, 14, 1898–1909. [Google Scholar] [CrossRef]
- Marteil, G.; Guerrero, A.; Vieira, A.F.; de Almeida, B.P.; Machado, P.; Mendonça, S.; Mesquita, M.; Villarreal, B.; Fonseca, I.; Francia, M.E.; et al. Over-elongation of centrioles in cancer promotes centriole amplification and chromosome missegregation. Nat. Commun. 2018, 9, 1258. [Google Scholar] [CrossRef]
- Lei, Q.; Xiong, L.; Xia, Y.; Feng, Z.; Gao, T.; Wei, W.; Song, X.; Ye, T.; Wang, N.; Peng, C.; et al. YLT-11, a novel PLK4 inhibitor, inhibits human breast cancer growth via inducing maladjusted centriole duplication and mitotic defect. Cell Death Dis. 2018, 9, 1066. [Google Scholar] [CrossRef]
- Kass, E.M.; Helgadottir, H.R.; Chen, C.C.; Barbera, M.; Wang, R.; Westermark, U.K.; Ludwig, T.; Moynahan, M.E.; Jasin, M. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 5564–5569. [Google Scholar] [CrossRef]
- Anestis, A.; Zoi, I.; Papavassiliou, A.G.; Karamouzis, M.V. Androgen Receptor in Breast Cancer-Clinical and Preclinical Research Insights. Molecules 2020, 25, 358. [Google Scholar] [CrossRef] [PubMed]
- Siddiqa, A.; Long, L.M.; Li, L.; Marciniak, R.A.; Kazhdan, I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer 2008, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Harari, D.; Yarden, Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 2000, 19, 6102–6114. [Google Scholar] [CrossRef]
- Kurokawa, H.; Lenferink, A.E.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000, 60, 5887–5894. [Google Scholar]
- Li, S.; Wu, C.; Fan, C.; Zhang, P.; Yu, G.; Li, K. Tanshinone II A improves the chemosensitivity of breast cancer cells to doxorubicin by inhibiting β-catenin nuclear translocation. J. Biochem. Mol. Toxicol. 2021, 35, e22620. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.S.; Oliveira, R.G.; Fraga, C.A.M.; Mermelstein, C.D.S.; Fernandes, P.D. ROCK inhibition with Fasudil induces beta-catenin nuclear translocation and inhibits cell migration of MDA-MB 231 human breast cancer cells. Sci. Rep. 2017, 7, 13723. [Google Scholar] [CrossRef]
- Ahram, M.; Bawadi, R.; Abdullah, M.S.; Alsafadi, D.B.; Abaza, H.; Abdallah, S.; Mustafa, E. Involvement of β-catenin in Androgen-induced Mesenchymal Transition of Breast MDA-MB-453 Cancer Cells. Endocr. Res. 2021, 46, 114–128. [Google Scholar] [CrossRef]
- Li, W.; Hou, J.Z.; Niu, J.; Xi, Z.Q.; Ma, C.; Sun, H.; Wang, C.J.; Fang, D.; Li, Q.; Xie, S.Q. Akt1 inhibition promotes breast cancer metastasis through EGFR-mediated β-catenin nuclear accumulation. Cell Commun. Signal 2018, 16, 82. [Google Scholar] [CrossRef]
- Manjer, J.; Johansson, R.; Berglund, G.; Janzon, L.; Kaaks, R.; Agren, A.; Lenner, P. Postmenopausal breast cancer risk in relation to sex steroid hormones, prolactin and SHBG (Sweden). Cancer Causes Control 2003, 14, 599–607. [Google Scholar] [CrossRef]
- Eliassen, A.H.; Missmer, S.A.; Tworoger, S.S.; Spiegelman, D.; Barbieri, R.L.; Dowsett, M.; Hankinson, S.E. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J. Natl. Cancer Inst. 2006, 98, 1406–1415. [Google Scholar] [CrossRef]
- Key, T.; Appleby, P.; Barnes, I.; Reeves, G. Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002, 94, 606–616. [Google Scholar] [CrossRef]
- Sasano, H.; Suzuki, T.; Miki, Y.; Moriya, T. Intracrinology of estrogens and androgens in breast carcinoma. J. Steroid Biochem. Mol. Biol. 2008, 108, 181–185. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Yeong, J.; Lim, J.C.T.; Lee, B.; Li, H.; Ong, C.C.H.; Thike, A.A.; Yeap, W.H.; Yang, Y.; Lim, A.Y.H.; Tay, T.K.Y.; et al. Prognostic value of CD8 + PD-1+ immune infiltrates and PDCD1 gene expression in triple negative breast cancer. J. Immunother. Cancer 2019, 7, 34. [Google Scholar] [CrossRef]
- Miao, Y.; Wang, J.; Li, Q.; Quan, W.; Wang, Y.; Li, C.; Wu, J.; Mi, D. Prognostic value and immunological role of PDCD1 gene in pan-cancer. Int. Immunopharmacol. 2020, 89 Pt B, 107080. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients with Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Adams, S.; Diamond, J.R.; Hamilton, E.P.; Pohlmann, P.R.; Tolaney, S.M.; Molinero, L.; He, X.; Waterkamp, D.; Funke, R.P.; Powderly, J.D. Phase Ib trial of atezolizumab in combination with nab-paclitaxel in patients with metastatic triple-negative breast cancer (mTNBC). J. Clin. Oncol. 2016, 34, 1009. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.B.; Blohmer, J.U.; Grischke, E.-M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; Rezai, M.; et al. A randomized phase II neoadjuvant study (GeparNuevo) to investigate the addition of durvalumab, a PD-L1 antibody, to a taxane-anthracycline containing chemotherapy in triple negative breast cancer (TNBC). J. Clin. Oncol. 2017, 35, 3062. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | ||||
|---|---|---|---|---|
| Variables | Total (n = 3060) | FHBC (n = 1676) | Non-FHBC (n = 1384) | p Value |
| n (%) | n (%) | n (%) | ||
| Biological factors | ||||
| Age (M ± SD) | 51.2 ± 8.2 | 49.7 ± 7.2 | 52.7 ± 9.1 | <0.001 |
| BMI (M ± SD) | 23.5 ± 3.7 | 23.5 ± 3.7 | 23.5 ± 3.7 | 0.994 |
| Age at menarche (M ± SD) | 13.3 ± 1.4 | 13.2 ±1.4 | 13.4 ± 1.5 | 0.040 |
| Age at 1st live birth (M ± SD) | 25.6 ± 6.1 | 24.5 ± 8.4 | 26.8 ±3.8 | <0.001 |
| First-degree relatives: | ||||
| Breast cancer mother | ||||
| No | 2230 (72.9) | 846 (27.6) | 1384 (45.2) | <0.001 |
| Yes | 830 (27.1) | 830 (27.1) | 0 (0.0) | |
| Breast cancer sisters | ||||
| No | 2185 (71.4) | 801 (26.2) | 1384 (45.2) | <0.001 |
| Yes | 875 (28.6) | 875 (28.6) | 0 (0.0) | |
| Has been pregnant | ||||
| No | 383 (12.5) | 212 (6.9) | 171 (5.6) | 0.821 |
| Yes | 2671 (87.5) | 1462 (47.9) | 1209 (39.6) | |
| Age at 1st pregnancy (M ± SD) | 25.6 ± 8.5 | 25.5 ± 4.5 | 25.6 ± 3.9 | 0.487 |
| Birth experience | ||||
| No | 468 (15.3) | 259 (8.5) | 209 (6.8) | 0.797 |
| Yes | 2585 (84.7) | 1414 (46.3) | 1171 (38.4) | |
| Breastfeeding practice | ||||
| Never | 260 (8.5) | 260 (8.5) | 0 (0.0) | <0.001 |
| No | 1089 (35.6) | 494 (16.1) | 595 (19.5) | |
| Yes | 1711 (55.9) | 922 (30.1) | 789 (25.8) | |
| Psychological and behavioral factors | ||||
| Depression status | ||||
| No | 2899 (94.9) | 1590 (52.1) | 1309 (42.8) | 0.945 |
| Yes | 156 (5.1) | 86 (2.8) | 70 (2.3) | |
| Alcohol consumption | ||||
| Never/occasionally | 2988 (97.6) | 1633 (53.4) | 1355 (44.3) | 0.011 |
| Not recently | 17 (0.6) | 5 (0.2) | 12 (0.4) | |
| Recently | 55 (1.8) | 38 (1.2) | 17 (0.6) | |
| Smoking | ||||
| No | 2760 (90.2) | 1490 (48.7) | 1270 (41.5) | 0.008 |
| Yes | 300 (9.8) | 186 (6.1) | 114 (3.7) | |
| Secondhand smoke | ||||
| No | 2842 (92.9) | 1548 (50.6) | 1294 (42.3) | 0.197 |
| Yes | 217 (4.1) | 128 (4.2) | 89 (2.9) | |
| Exercise | ||||
| No | 1783 (58.3) | 1011 (33.1) | 772 (25.2) | 0.011 |
| Yes | 1276 (41.7) | 664 (21.7) | 612 (20.0) | |
| Social factors | ||||
| Marital Status | ||||
| Unmarried | 344 (11.3) | 197 (6.4) | 147 (4.8) | 0.334 |
| Married | 2713 (88.7) | 1479 (48.4) | 1234 (40.4) | |
| Educational level | ||||
| Primary school | 337 (11.0) | 180 (5.9) | 157 (5.1) | 0.590 |
| High school or above | 2722 (89.0) | 1496 (48.9) | 1226 (40.1) | |
| Job experience | ||||
| No | 2160 (70.6) | 1286 (42.0) | 874 (28.6) | <0.001 |
| Yes | 900 (29.4) | 390 (12.7) | 510 (16.7) | |
| Live alone | ||||
| No | 2777 (90.8) | 1540 (50.3) | 1237 (40.4) | 0.020 |
| Yes | 282 (9.2) | 136 (4.4) | 146 (4.8) | |
| No | Potential Genes | DEMs | DEmiRNAs | |||||
|---|---|---|---|---|---|---|---|---|
| CpG sites | Location | p Value | FDR | hsa-miRNA | p Value | FDR | ||
| 1 | GNPDA1 * | cg22647996 | TSS1500 | 0.000476001 | 0.639025617 | hsa-miR-23a-3p | 5.88 × 10−37 | 1.19 × 10−34 |
| 2 | ADARB2 | cg20205188 | Body | 0.000931185 | 0.891526514 | hsa-miR-654-3p | 2.51 × 10−19 | 1.70 × 10−17 |
| 3 | STOX2 | cg27457427 | Body | 0.0000935 | 0.255342042 | hsa-miR-22-3p | 9.18 × 10−20 | 6.66 × 10−18 |
| 4 | SHROOM2 | cg23553400 | Body | 0.000206531 | 0.4024522855 | hsa-miR-23a-3p | 5.88 × 10−37 | 1.19 × 10−34 |
| 5 | NCAPD3 | cg14934141 | Body | 0.000345934 | 0.542808906 | hsa-miR-654-3p | 2.51 × 10−19 | 1.70 × 10−17 |
| 6 | CCKBR | cg26313599 | Body | 0.000435502 | 0.613241497 | hsa-miR-654-3p | 2.51 × 10−19 | 1.70 × 10−17 |
| 7 | ADAM19 | cg19464247 | Body | 0.005936274 | 1 | hsa-miR-23a-3p | 5.88 × 10−37 | 1.19 × 10−34 |
| 8 | SLC25A16 * | cg26546862 | TSS1500 | 0.004554406 | 1 | hsa-miR-425-5p | 3.79 × 10−38 | 9.63 × 10−36 |
| 9 | DLGAP2 | cg21498547 | 3′UTR | 0.011187133 | 1 | hsa-miR-22-3p | 9.18 × 10−20 | 6.66 × 10−18 |
| 10 | ZNF787 | cg26951705 | Body | 0.017288423 | 1 | hsa-miR-423-5p | 8.53 × 10−19 | 4.81 × 10−17 |
| 11 | MRT04 | cg05704942 | Body | 0.046991025 | 1 | hsa-miR-654-3p | 2.51 × 10−19 | 1.70 × 10−17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khairi, S.; Wang, C.-Y.; Anuraga, G.; Prayugo, F.B.; Ansar, M.; Lesmana, M.H.S.; Irham, L.M.; Shen, C.-Y.; Chung, M.-H. Integrative Analysis of DNA Methylation and microRNA Reveals GNPDA1 and SLC25A16 Related to Biopsychosocial Factors Among Taiwanese Women with a Family History of Breast Cancer. J. Pers. Med. 2025, 15, 134. https://doi.org/10.3390/jpm15040134
Khairi S, Wang C-Y, Anuraga G, Prayugo FB, Ansar M, Lesmana MHS, Irham LM, Shen C-Y, Chung M-H. Integrative Analysis of DNA Methylation and microRNA Reveals GNPDA1 and SLC25A16 Related to Biopsychosocial Factors Among Taiwanese Women with a Family History of Breast Cancer. Journal of Personalized Medicine. 2025; 15(4):134. https://doi.org/10.3390/jpm15040134
Chicago/Turabian StyleKhairi, Sabiah, Chih-Yang Wang, Gangga Anuraga, Fidelia Berenice Prayugo, Muhamad Ansar, Mohammad Hendra Setia Lesmana, Lalu Muhammad Irham, Chen-Yang Shen, and Min-Huey Chung. 2025. "Integrative Analysis of DNA Methylation and microRNA Reveals GNPDA1 and SLC25A16 Related to Biopsychosocial Factors Among Taiwanese Women with a Family History of Breast Cancer" Journal of Personalized Medicine 15, no. 4: 134. https://doi.org/10.3390/jpm15040134
APA StyleKhairi, S., Wang, C.-Y., Anuraga, G., Prayugo, F. B., Ansar, M., Lesmana, M. H. S., Irham, L. M., Shen, C.-Y., & Chung, M.-H. (2025). Integrative Analysis of DNA Methylation and microRNA Reveals GNPDA1 and SLC25A16 Related to Biopsychosocial Factors Among Taiwanese Women with a Family History of Breast Cancer. Journal of Personalized Medicine, 15(4), 134. https://doi.org/10.3390/jpm15040134