

Laboratory Tools to Predict CFTR Modulator Therapy Effectiveness and to Monitor Disease Severity in Cystic Fibrosis


Abstract

1. Introduction
2. Laboratory Tools to Predict CFTR Modulator Effectiveness In Vivo
2.1. Primary Airway Cells Grown in Monolayers
2.2. Airway Organoids/Nasospheroids
2.3. Intestinal Organoids
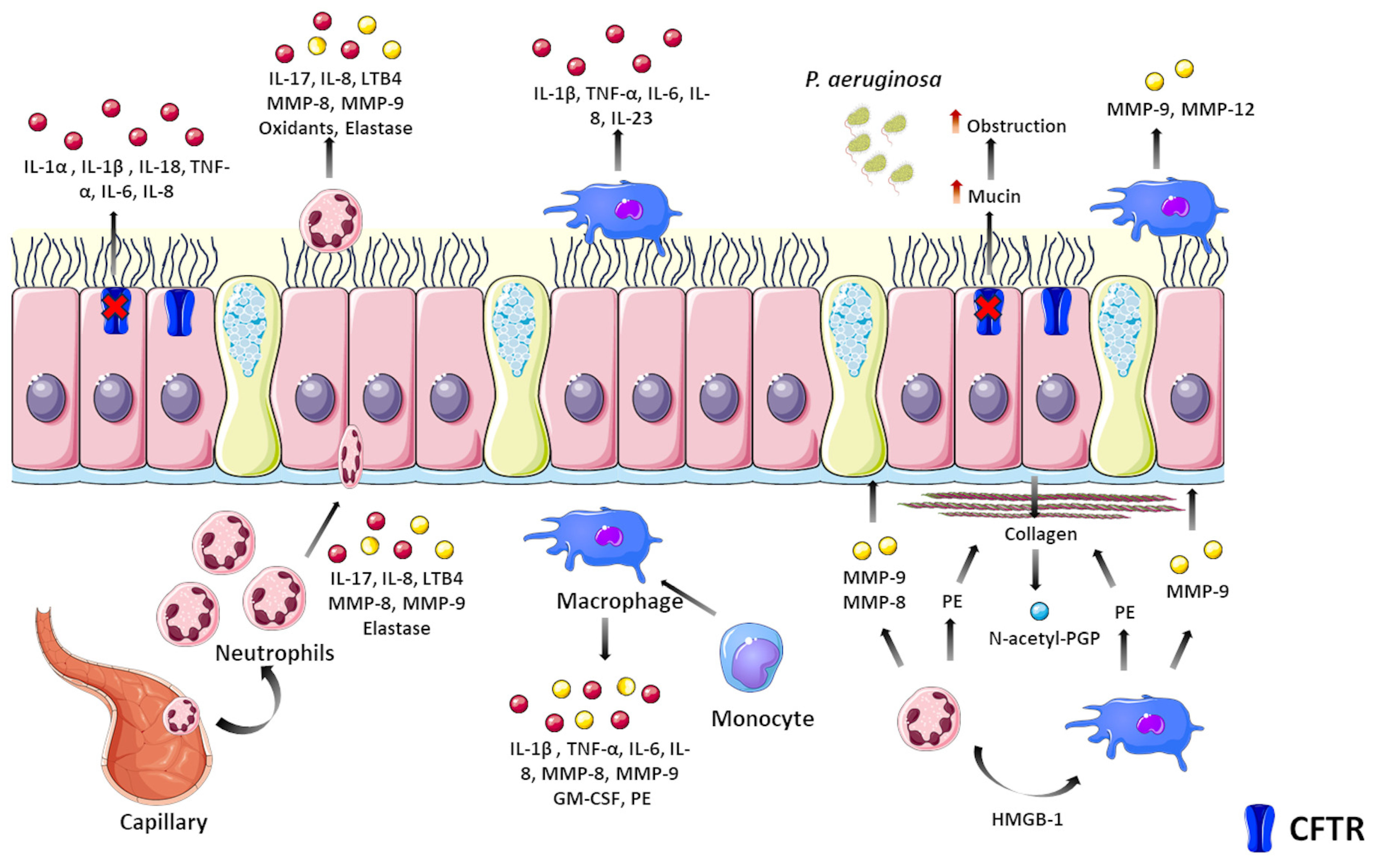
3. Laboratory Biomarkers to Monitor Lung Disease Progression
3.1. Monitoring Ivacaftor Effects on CF Inflammation
3.2. Monitoring Lumacaftor/Ivacaftor and Tezacaftor/Ivacaftor Effects on CF Inflammation
3.3. Monitoring Elexacaftor/Tezacaftor/Ivacaftor Effects on CF Inflammation
4. Novel Laboratory Tools to Assess CF Status and Progression
4.1. Extracellular Vesicles
4.2. MicroRNAs
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Garratt, A.; Hill, A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J. Cyst. Fibros. 2022, 21, 456–462. [Google Scholar] [CrossRef]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef] [PubMed]
- Boon, M.; Verleden, S.E.; Bosch, B.; Lammertyn, E.J.; McDonough, J.E.; Mai, C.; Verschakelen, J.; Kemner-van de Corput, M.; Tiddens, H.A.W.; Proesmans, M.; et al. Morphometric Analysis of Explant Lungs in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 516–526. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef]
- Sosnay, P.R.; White, T.B.; Farrell, P.M.; Ren, C.L.; Derichs, N.; Howenstine, M.S.; Nick, J.A.; De Boeck, K. Diagnosis of Cystic Fibrosis in Nonscreened Populations. J. Pediatr. 2017, 181S, S52–S57.e2. [Google Scholar] [CrossRef]
- Cystic Fibrosis Mutation Database (CFTR1 Database). Available online: http://www.genet.sickkids.on.ca/ (accessed on 1 December 2023).
- Clinical and Function Translation of CFTR (CFTR2 Database). Available online: https://cftr2.org/ (accessed on 1 December 2023).
- Koch, C.; Cuppens, H.; Rainisio, M.; Madessani, U.; Harms, H.; Hodson, M.; Mastella, G.; Navarro, J.; Strandvik, B.; McKenzie, S.; et al. European Epidemiologic Registry of Cystic Fibrosis (ERCF): Comparison of major disease manifestations between patients with different classes of mutations. Pediatr. Pulmonol. 2001, 31, 1–12. [Google Scholar] [CrossRef]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef]
- Oliver, K.E.; Carlon, M.S.; Pedemonte, N.; Lopes-Pacheco, M. The revolution of personalized pharmacotherapies for cystic fibrosis: What does the future hold? Expert Opin. Pharmacother. 2023, 24, 1545–1565. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.C.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Bacalhau, M.; Camargo, M.; Magalhães-Ghiotto, G.A.V.; Drumond, S.; Castelletti, C.H.M.; Lopes-Pacheco, M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals 2023, 16, 410. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 375. [Google Scholar] [CrossRef] [PubMed]
- Guimbellot, J.S.; Leach, J.M.; Chaudhry, I.G.; Quinney, N.L.; Boyles, S.E.; Chua, M.; Aban, I.; Jaspers, I.; Gentzsch, M. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017, 2, e95734. [Google Scholar] [CrossRef]
- Schork, N.J. Personalized medicine: Time for one-person trials. Nature 2015, 520, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Vohra, S.; Shamseer, L.; Sampson, M.; Bukutu, C.; Schmid, C.H.; Tate, R.; Nikles, J.; Zucker, D.R.; Kravitz, R.; Guyatt, G.; et al. CONSORT extension for reporting N-of-1 trials (CENT) 2015 Statement. J. Clin. Epidemiol. 2016, 76, 9–17. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Hinzpeter, A.; Caci, E.; Pedemonte, N.; Arous, N.; Di Duca, M.; Zegarra-Moran, O.; Fanen, P.; Galietta, L.J.V. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J. Pharmacol. Exp. Ther. 2009, 330, 786–791. [Google Scholar] [CrossRef]
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef]
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; De Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748. [Google Scholar] [CrossRef]
- Fidler, M.C.; Buckley, A.; Sullivan, J.C.; Statia, M.; Boj, S.F.; Vries, R.G.J.; Munck, A.; Higgins, M.; Moretto Zita, M.; Negulescu, P.; et al. G970R-CFTR Mutation (c.2908G>C) Results Predominantly in a Splicing Defect. Clin. Transl. Sci. 2021, 14, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J.V. Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol. Cell Physiol. 2010, 298, C866–C874. [Google Scholar] [CrossRef] [PubMed]
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J.V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Cell Physiol. 2011, 301, C872–C885. [Google Scholar] [CrossRef] [PubMed]
- Tomati, V.; Costa, S.; Capurro, V.; Pesce, E.; Pastorino, C.; Lena, M.; Sondo, E.; Di Duca, M.; Cresta, F.; Cristadoro, S.; et al. Rescue by elexacaftor-tezacaftor-ivacaftor of the G1244E cystic fibrosis mutation’s stability and gating defects are dependent on cell background. J. Cyst. Fibros. 2023, 22, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Sepahzad, A.; Morris-Rosendahl, D.J.; Davies, J.C. Cystic fibrosis lung disease modifiers and their relevance in the new era of precision medicine. Genes 2021, 12, 562. [Google Scholar] [CrossRef]
- McGarry, M.E.; McColley, S.A. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr. Pulmonol. 2021, 56, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018, 3, e99385. [Google Scholar] [CrossRef]
- Noel, S.; Servel, N.; Hatton, A.; Golec, A.; Rodrat, M.; Ng, D.R.S.; Li, H.; Pranke, I.; Hinzpeter, A.; Edelman, A.; et al. Correlating genotype with phenotype using CFTR-mediated whole-cell Cl− currents in human nasal epithelial cells. J. Physiol. 2022, 600, 1515–1531. [Google Scholar] [CrossRef]
- Debley, J.S.; Barrow, K.A.; Rich, L.M.; Singh, P.; McKone, E.F.; Nichols, D.P. Correlation between ivacaftor-induced CFTR activation in airway epithelial cells and improved lung function: A proof-of-concept study. Ann. Am. Thorac. Soc. 2020, 17, 1024–1027. [Google Scholar] [CrossRef]
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Olshansky, S.; Moreno, C.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479. [Google Scholar] [CrossRef]
- Park, J.K.H.; Shrivastava, A.; Zhang, C.; Pollok, B.A.; Finkbeiner, W.E.; Gibb, E.R.; Ly, N.P.; Illek, B. Functional Profiling of CFTR-Directed Therapeutics Using Pediatric Patient-Derived Nasal Epithelial Cell Models. Front. Pediatr. 2020, 8, 536. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Pesce, E.; Capurro, V.; Tomati, V.; Lena, M.; Pastorino, C.; Bocciardi, R.; Zara, F.; Centrone, C.; Taccetti, G.; et al. Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators. Int. J. Mol. Sci. 2023, 24, 6576. [Google Scholar] [CrossRef] [PubMed]
- Dreano, E.; Burgel, P.R.; Hatton, A.; Bouazza, N.; Chevalier, B.; Macey, J.; Leroy, S.; Durieu, I.; Weiss, L.; Grenet, D.; et al. Theratyping cystic fibrosis patients to guide elexacaftor/tezacaftor/ivacaftor out-of-label prescription. Eur. Respir. J. 2023, 62, 2300110. [Google Scholar] [CrossRef] [PubMed]
- Allan, K.M.; Astore, M.A.; Kardia, E.; Wong, S.L.; Fawcett, L.K.; Bell, J.L.; Visser, S.; Chen, P.-C.; Griffith, R.; Jaffe, A.; et al. Q1291H-CFTR molecular dynamics simulations and ex vivo theratyping in nasal epithelial models and clinical response to elexacaftor/tezacaftor/ivacaftor in a Q1291H/F508del patient. Front. Mol. Biosci. 2023, 10, 1148501. [Google Scholar] [CrossRef] [PubMed]
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Ostmann, A.J.; Strecker, L.M.; Szczesniak, R.D.; Clancy, J.P. Detection of CFTR function and modulation in primary human nasal cell spheroids. J. Cyst. Fibros. 2018, 17, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.D.; Liu, Z.; Odom, L.V.; Kersh, L.; Guimbellot, J.S. CFTR function and clinical response to modulators parallel nasal epithelial organoid swelling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L119–L129. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708. [Google Scholar] [CrossRef]
- de Winter-De Groot, K.M.; Janssens, H.M.; van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur. Respir. J. 2018, 52, 1702529. [Google Scholar] [CrossRef]
- de Winter-de Groot, K.M.; Berkers, G.; Marck-van der Wilt, R.E.P.; van der Meer, R.; Vonk, A.; Dekkers, J.F.; Geerdink, M.; Michel, S.; Kruisselbrink, E.; Vries, R.; et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J. Cyst. Fibros. 2020, 19, 614–619. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Förstová, E.; Vonk, A.M.; Ferrante, M.; Verfailli, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekma, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef] [PubMed]
- Aalbers, B.L.; Brunsveld, J.E.; van der Ent, C.K.; van den Eijnden, J.C.; Beekman, J.M.; Heijerman, H.G.M. Forskolin induced swelling (FIS) assay in intestinal organoids to guide eligibility for compassionate use treatment in a CF patient with a rare genotype. J. Cyst. Fibros. 2022, 21, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of organoid swelling and in vivo biomarkers of cftr function to determine effects of lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for the f508del mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- Muilwijk, D.; de Poel, E.; van Mourik, P.; Suen, S.W.F.; Vonk, A.M.; Brunsveld, J.E.; Kruisselbrink, E.; Oppelaar, H.; Hagemeijer, M.C.; Berkers, G.; et al. Forskolin-induced Organoid Swelling is Associated with Long-term CF Disease Progression. Eur. Respir. J. 2022, 60, 2100508. [Google Scholar] [CrossRef]
- Lefferts, J.W.; Bierlaagh, M.C.; Kroes, S.; Nieuwenhuijze, N.D.A.; Sonneveld van Kooten, H.N.; Niemöller, P.J.; Verburg, T.F.; Janssens, H.M.; Muilwijk, D.; van Beuningen, S.F.B.; et al. CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes. Int. J. Mol. Sci. 2023, 24, 14539. [Google Scholar] [CrossRef]
- Mitropoulou, G.; Brandenberg, N.; Hoehnel, S.; Ceroni, C.; Balmpouzis, Z.; Blanchon, S.; Dorta, G.; Sauty, A.; Koutsokera, A. Rectal organoid-guided CFTR modulator therapy restores lung function in a CF patient with the rare 1677delTA/R334W genotype. Eur. Respir. J. 2022, 60, 2201341. [Google Scholar] [CrossRef]
- Kondratyeva, E.; Melyanovskaya, Y.; Efremova, A.; Krasnova, M.; Mokrousova, D.; Bulatenko, N.; Petrova, N.; Polyakov, A.; Adyan, T.; Kovalskaia, V.; et al. Clinical and Genetic Characteristics of a Patient with Cystic Fibrosis with a Complex Allele [E217G;G509D] and Functional Evaluation of the CFTR Channel. Genes 2023, 14, 1705. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Fulcher, M.L.; Randell, S.H. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol. Biol. 2013, 945, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef]
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.H.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 2016, 19, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef]
- Müller, L.; Brighton, L.E.; Carson, J.L.; Fischer, W.A.; Jaspers, I. Culturing of human nasal epithelial cells at the air liquid interface. J. Vis. Exp. 2013, 80, e50646. [Google Scholar] [CrossRef]
- Sette, G.; Lo Cicero, S.; Blaconà, G.; Pierandrei, S.; Bruno, S.M.; Salvati, V.; Castelli, G.; Falchi, M.; Fabrizzi, B.; Cimino, G.; et al. Theratyping cystic fibrosis in vitro in ALI culture and organoid models generated from patient-derived nasal epithelial conditionally reprogrammed stem cells. Eur. Respir. J. 2021, 58, 2100908. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.; Li, M.; Li, J.; Shen, J.; Zhao, Y.; Pang, J.; Wen, Q.; Chen, M.; Wei, B.; et al. Conditional reprogramming: Next generation cell culture. Acta Pharm. Sin. B 2020, 10, 1360–1381. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef]
- McDougall, C.M.; Blaylock, M.G.; Douglas, J.G.; Brooker, R.J.; Helms, P.J.; Walsh, G.M. Nasal epithelial cells as surrogates for bronchial epithelial cells in airway inflammation studies. Am. J. Respir. Cell Mol. Biol. 2008, 39, 560–568. [Google Scholar] [CrossRef]
- Li, H.; Sheppard, D.N.; Hug, M.J. Transepithelial electrical measurements with the Ussing chamber. J. Cyst. Fibros. 2004, 3 (Suppl. S2), 123–126. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.N.; Gray, M.A.; Gong, X.; Sohma, Y.; Kogan, I.; Benos, D.J.; Scott-Ward, T.S.; Chen, J.-H.; Li, H.; Cai, Z.; et al. The patch-clamp and planar lipid bilayer techniques: Powerful and versatile tools to investigate the CFTR Cl− channel. J. Cyst. Fibros. 2004, 3, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Balázs, A.; Ziegahn, N.; Rubil, T.; Vitzthum, C.; Piehler, L.; Drescher, M.; Seidel, K.; Rohrbach, A.; Röhmel, J.; et al. Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis. Int. J. Mol. Sci. 2023, 24, 12365. [Google Scholar] [CrossRef]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might brushed nasal cells be a surrogate for CFTR modulator clinical response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Calucho, M.; Gartner, S.; Barranco, P.; Fernández-Álvarez, P.; Pérez, R.G.; Tizzano, E.F. Validation of nasospheroids to assay CFTR functionality and modulator responses in cystic fibrosis. Sci. Rep. 2021, 11, 15511. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Anderson, J.D.; Deng, L.; Mackay, S.; Bailey, J.; Kersh, L.; Rowe, S.M.; Guimbellot, J.S. Human nasal epithelial organoids for therapeutic development in cystic fibrosis. Genes 2020, 11, 603. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-De Groot, K.M.; Brandsma, A.M.; De Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mall, M. Electrolyte transport in the mammalian colon: Mechanisms and implications for disease. Physiol. Rev. 2002, 82, 245–289. [Google Scholar] [CrossRef] [PubMed]
- Cuyx, S.; Ramalho, A.S.; Corthout, N.; Fieuws, S.; Fürstová, E.; Arnauts, K.; Ferrante, M.; Verfaillie, C.; Munck, S.; Boon, M.; et al. Rectal organoid morphology analysis (ROMA) as a promising diagnostic tool in cystic fibrosis. Thorax 2021, 76, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef]
- Tiddens, H.A.W.M.; Puderbach, M.; Venegas, J.G.; Ratjen, F.; Donaldson, S.H.; Davis, S.D.; Rowe, S.M.; Sagel, S.D.; Higgins, M.; Waltz, D.A. Novel outcome measures for clinical trials in cystic fibrosis. Pediatr. Pulmonol. 2015, 50, 302–315. [Google Scholar] [CrossRef]
- Kyrilli, S.; Henry, T.; Wilschanski, M.; Fajac, I.; Davies, J.C.; Jais, J.P.; Sermet-Gaudelus, I. Insights into the variability of nasal potential difference, a biomarker of CFTR activity. J. Cyst. Fibros. 2020, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Fidler, M.C.; Beusmans, J.; Panorchan, P.; Van Goor, F. Correlation of sweat chloride and percent predicted FEV1 in cystic fibrosis patients treated with ivacaftor. J. Cyst. Fibros. 2017, 16, 41–44. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hanrahan, J.W.; Bilodeau, G.; Ellis, L.; Dupuis, A.; Liao, J.; Zielenski, J.; Durie, P. Cystic Fibrosis Transmembrane Conductance Regulator Function Is Suppressed in Cigarette Smokers. Am. J. Respir. Crit. Care Med. 2006, 173, 1139–1144. [Google Scholar] [CrossRef]
- Koerbin, G.; Greaves, R.F.; Robins, H.; Farquhar, J.; Hickman, P.E. Total intra-individual variation in sweat sodium and chloride concentrations for the diagnosis of cystic fibrosis. Clin. Chim. Acta 2008, 393, 128–129. [Google Scholar] [CrossRef]
- Vermeulen, F.; Lebecque, P.; De Boeck, K.; Leal, T. Biological variability of the sweat chloride in diagnostic sweat tests: A retrospective analysis. J. Cyst. Fibros. 2017, 16, 30–35. [Google Scholar] [CrossRef]
- Nichols, D.; Chmiel, J.; Berger, M. Chronic Inflammation in the Cystic Fibrosis Lung: Alterations in Inter- and Intracellular Signaling. Clin. Rev. Allergy Immunol. 2008, 34, 146–162. [Google Scholar] [CrossRef]
- Carbone, A.; Vitullo, P.; Di Gioia, S.; Conese, M. Lung Inflammatory Genes in Cystic Fibrosis and Their Relevance to Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapies. Genes 2023, 14, 1966. [Google Scholar] [CrossRef]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical Mechanism of the Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor in G551D-mediated Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Bernarde, C.; Keravec, M.; Mounier, J.; Gouriou, S.; Rault, G.; Férec, C.; Barbier, G.; Héry-Arnaud, G. Impact of the CFTR-Potentiator Ivacaftor on Airway Microbiota in Cystic Fibrosis Patients Carrying A G551D Mutation. PLoS ONE 2015, 10, e0124124. [Google Scholar] [CrossRef]
- Guerra, L.; D’Oria, S.; Favia, M.; Castellani, S.; Santostasi, T.; Polizzi, A.M.; Mariggiò, M.A.; Gallo, C.; Casavola, V.; Montemurro, P.; et al. CFTR-dependent chloride efflux in cystic fibrosis mononuclear cells is increased by ivacaftor therapy. Pediatr. Pulmonol. 2017, 52, 900–908. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Ronan, N.J.; Einarsson, G.G.; Twomey, M.; Mooney, D.; Mullane, D.; NiChroinin, M.; O’Callaghan, G.; Shanahan, F.; Murphy, D.M.; O’Connor, O.J.; et al. CORK Study in Cystic Fibrosis: Sustained Improvements in Ultra-Low-Dose Chest CT Scores after CFTR Modulation with Ivacaftor. Chest 2018, 153, 395–403. [Google Scholar] [CrossRef]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Mainz, J.G.; Arnold, C.; Wittstock, K.; Hipler, U.-C.; Lehmann, T.; Zagoya, C.; Duckstein, F.; Ellemunter, H.; Hentschel, J. Ivacaftor Reduces Inflammatory Mediators in Upper Airway Lining Fluid From Cystic Fibrosis Patients With a G551D Mutation: Serial Non-Invasive Home-Based Collection of Upper Airway Lining Fluid. Front. Immunol. 2021, 12, 642180. [Google Scholar] [CrossRef]
- Wilson, J.; Keating, D.; Clark, D.; Edgeworth, D.; Allen-Graham, J.; Finlayson, F.; Mifsud, N.; Williams, E.; Kotsimbos, T. 73 The effect of ivacaftor CFTR gene-potentiating therapy on cytokine levels in CF patients with the G551D mutation. J. Cyst. Fibros. 2017, 16, S83. [Google Scholar] [CrossRef]
- White, M.M.; Geraghty, P.; Hayes, E.; Cox, S.; Leitch, W.; Alfawaz, B.; Lavelle, G.M.; McElvaney, O.J.; Flannery, R.; Keenan, J.; et al. Neutrophil Membrane Cholesterol Content is a Key Factor in Cystic Fibrosis Lung Disease. eBioMedicine 2017, 23, 173–184. [Google Scholar] [CrossRef]
- McNally, P.; Butler, D.; Karpievitch, Y.V.; Linnane, B.; Ranganathan, S.; Stick, S.M.; Hall, G.L.; Schultz, A. Ivacaftor and Airway Inflammation in Preschool Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Favia, M.; Gallo, C.; Guerra, L.; De Venuto, D.; Diana, A.; Polizzi, A.M.; Montemurro, P.; Mariggiò, M.A.; Leonetti, G.; Manca, A.; et al. Treatment of Cystic Fibrosis Patients Homozygous for F508del with Lumacaftor-Ivacaftor (Orkambi®) Restores Defective CFTR Channel Function in Circulating Mononuclear Cells. Int. J. Mol. Sci. 2020, 21, 2398. [Google Scholar] [CrossRef] [PubMed]
- Arooj, P.; Morrissy, D.V.; McCarthy, Y.; Vagg, T.; McCarthy, M.; Fleming, C.; Daly, M.; Eustace, J.A.; Murphy, D.M.; Plant, B.J. ROCK STUDY in CF: Sustained anti-inflammatory effects of lumacaftor–ivacaftor in sputum and peripheral blood samples of adult patients with cystic fibrosis—An observational study. BMJ Open Respir. Res. 2023, 10, e001590. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Boutin, S.; Wielpütz, M.O.; Joachim, C.; Frey, D.L.; Wege, S.; Sommerburg, O.; Kauczor, H.-U.; Stahl, M.; Dalpke, A.H.; et al. Effects of Lumacaftor-Ivacaftor on Lung Clearance Index, Magnetic Resonance Imaging, and Airway Microbiome in Phe508del Homozygous Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2021, 18, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Enaud, R.; Lussac-Sorton, F.; Charpentier, E.; Velo-Suárez, L.; Guiraud, J.; Bui, S.; Fayon, M.; Schaeverbeke, T.; Nikolski, M.; Burgel, P.-R.; et al. Effects of Lumacaftor-Ivacaftor on Airway Microbiota-Mycobiota and Inflammation in Patients with Cystic Fibrosis Appear To Be Linked to Pseudomonas aeruginosa Chronic Colonization. Microbiol. Spectr. 2023, 11, e02251-22. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Griffiths, H.H.; Scambler, T.; Wong, C.H.; Lara-Reyna, S.; Holbrook, J.; Martinon, F.; Savic, S.; Whitaker, P.; Etherington, C.; Spoletini, G.; et al. Different CFTR modulator combinations downregulate inflammation differently in cystic fibrosis. eLife 2020, 9, e54556. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.; Britt, R.D.; Ryan-Wenger, N.A.; Khan, A.Q.; Lewis, B.W.; Gushue, C.; Ozuna, H.; Jaganathan, D.; McCoy, K.; Kopp, B.T. Impact of elexacaftor-tezacaftor-ivacaftor on bacterial colonization and inflammatory responses in cystic fibrosis. Pediatr. Pulmonol. 2023, 58, 825–833. [Google Scholar] [CrossRef]
- Schaupp, L.; Addante, A.; Völler, M.; Fentker, K.; Kuppe, A.; Bardua, M.; Duerr, J.; Piehler, L.; Röhmel, J.; Thee, S.; et al. Longitudinal effects of elexacaftor/tezacaftor/ivacaftor on sputum viscoelastic properties, airway infection and inflammation in patients with cystic fibrosis. Eur. Respir. J. 2023, 62, 2202153. [Google Scholar] [CrossRef]
- De Vuyst, R.C.; Bennard, E.; Kam, C.W.; McKinzie, C.J.; Esther, C.R. Elexacaftor/tezacaftor/ivacaftor treatment reduces airway inflammation in cystic fibrosis. Pediatr. Pulmonol. 2023, 58, 1592–1594. [Google Scholar] [CrossRef]
- Casey, M.; Gabillard-Lefort, C.; McElvaney, O.F.; McElvaney, O.J.; Carroll, T.; Heeney, R.C.; Gunaratnam, C.; Reeves, E.P.; Murphy, M.P.; McElvaney, N.G. Effect of elexacaftor/tezacaftor/ivacaftor on airway and systemic inflammation in cystic fibrosis. Thorax 2023, 78, 835–839. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, S.M. Pseudomonas aeruginosa in Cystic Fibrosis Patients with G551D-CFTR Treated with Ivacaftor. Clin. Infect. Dis. 2015, 60, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, J.E.; Wagner, B.D.; Kirk Harris, J.; Rowe, S.M.; Heltshe, S.L.; DeBoer, E.M.; Sagel, S.D. Effects of ivacaftor on systemic inflammation and the plasma proteome in people with CF and G551D. J. Cyst. Fibros. 2022, 21, 950–958. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef]
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfsen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J. Cyst. Fibros. 2019, 18, 102–109. [Google Scholar] [CrossRef]
- Salvatore, D.; Carnovale, V.; Iacotucci, P.; Braggion, C.; Castellani, C.; Cimino, G.; Colangelo, C.; Francalanci, M.; Leonetti, G.; Lucidi, V.; et al. Effectivenesss of ivacaftor in severe cystic fibrosis patients and non-G551D gating mutations. Pediatr. Pulmonol. 2019, 54, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The impact of FDA and EMA regulatory decision—Making process on the access to CFTR modulators for the treatment of cystic fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef]
- Sagel, S.D.; Chmiel, J.F.; Konstan, M.W. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proc. Am. Thorac. Soc. 2007, 4, 406–417. [Google Scholar] [CrossRef]
- Fayon, M.; Kent, L.; Bui, S.; Dupont, L.; Sermet, I.; European Cystic Fibrosis Society Clinical Trial Network Standardisation Committee. Clinimetric properties of bronchoalveolar lavage inflammatory markers in cystic fibrosis. Eur. Respir. J. 2014, 43, 610–626. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Aitken, M.L.; Accurso, F.J.; Kronmal, R.A.; Konstan, M.W.; Burns, J.L.; Sagel, S.D.; Ramsey, B.W. Association between Pulmonary Function and Sputum Biomarkers in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 822–828. [Google Scholar] [CrossRef]
- Lepissier, A.; Addy, C.; Hayes, K.; Noel, S.; Bui, S.; Burgel, P.-R.; Dupont, L.; Eickmeier, O.; Fayon, M.; Leal, T.; et al. Inflammation biomarkers in sputum for clinical trials in cystic fibrosis: Current understanding and gaps in knowledge. J. Cyst. Fibros. 2022, 21, 691–706. [Google Scholar] [CrossRef]
- Abreu, S.C.; Lopes-Pacheco, M.; Weiss, D.J.; Rocco, P.R.M. Mesenchymal Stromal Cell-Derived Extracellular Vesicles in Lung Diseases: Current Status and Perspectives. Front. Cell Dev. Biol. 2021, 9, 600711. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.C.; Lopes-Pacheco, M.; English, K.; Rolandsson Enes, S.; Krasnodembskaya, A.; Rocco, P.R.M. The MSC-EV-microRNAome: A Perspective on Therapeutic Mechanisms of Action in Sepsis and ARDS. Cells 2024, 13, 122. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, S.; Daniello, V.; Conese, M. Extracellular Vesicles’ Role in the Pathophysiology and as Biomarkers in Cystic Fibrosis and COPD. Int. J. Mol. Sci. 2022, 24, 228. [Google Scholar] [CrossRef] [PubMed]
- Letsiou, E.; Bauer, N. Endothelial Extracellular Vesicles in Pulmonary Function and Disease. In Current Topics in Membranes; Academic Press: Cambridge, MA, USA, 2018; Volume 82, pp. 197–256. [Google Scholar]
- Fujita, Y.; Kadota, T.; Araya, J.; Ochiya, T.; Kuwano, K. Extracellular Vesicles: New Players in Lung Immunity. Am. J. Respir. Cell Mol. Biol. 2018, 58, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Haggadone, M.D.; Peters-Golden, M. Microenvironmental Influences on Extracellular Vesicle-Mediated Communication in the Lung. Trends Mol. Med. 2018, 24, 963–975. [Google Scholar] [CrossRef]
- Su, G.; Ma, X.; Wei, H. Multiple Biological Roles of Extracellular Vesicles in Lung Injury and Inflammation Microenvironment. BioMed Res. Int. 2020, 2020, 5608382. [Google Scholar] [CrossRef]
- Porro, C.; Lepore, S.; Trotta, T.; Castellani, S.; Ratclif, L.; Battaglino, A.; Di Gioia, S.; Martínez, M.C.; Conese, M.; Maffione, A.B. Isolation and characterization of microparticles in sputum from cystic fibrosis patients. Respir. Res. 2010, 11, 94. [Google Scholar] [CrossRef]
- Kadota, T.; Fujita, Y.; Yoshioka, Y.; Araya, J.; Kuwano, K.; Ochiya, T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2016, 17, 1801. [Google Scholar] [CrossRef]
- Trappe, A.; Donnelly, S.C.; McNally, P.; Coppinger, J.A. Role of extracellular vesicles in chronic lung disease. Thorax 2021, 76, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Di Gioia, S.; Trotta, T.; Lepore, S.; Panaro, M.A.; Battaglino, A.; Ratclif, L.; Castellani, S.; Bufo, P.; Martinez, M.C.; et al. Pro-inflammatory effect of cystic fibrosis sputum microparticles in the murine lung. J. Cyst. Fibros. 2013, 12, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Rollet-Cohen, V.; Bourderioux, M.; Lipecka, J.; Chhuon, C.; Jung, V.A.; Mesbahi, M.; Nguyen-Khoa, T.; Guérin-Pfyffer, S.; Schmitt, A.; Edelman, A.; et al. Comparative proteomics of respiratory exosomes in cystic fibrosis, primary ciliary dyskinesia and asthma. J. Proteom. 2018, 185, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Useckaite, Z.; Ward, M.P.; Trappe, A.; Reilly, R.; Lennon, J.; Davage, H.; Matallanas, D.; Cassidy, H.; Dillon, E.T.; Brennan, K.; et al. Increased extracellular vesicles mediate inflammatory signalling in cystic fibrosis. Thorax 2020, 75, 449–458. [Google Scholar] [CrossRef]
- Forrest, O.A.; Dobosh, B.; Ingersoll, S.A.; Rao, S.; Rojas, A.; Laval, J.; Alvarez, J.A.; Brown, M.R.; Tangpricha, V.; Tirouvanziam, R. Neutrophil-derived extracellular vesicles promote feed-forward inflammasome signaling in cystic fibrosis airways. J. Leukoc. Biol. 2022, 112, 707–716. [Google Scholar] [CrossRef]
- Trappe, A.; Lakkappa, N.; Carter, S.; Dillon, E.; Wynne, K.; McKone, E.; McNally, P.; Coppinger, J.A. Investigating serum extracellular vesicles in Cystic Fibrosis. J. Cyst. Fibros. 2023, 22, 674–679. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef]
- Krause, K.; Kopp, B.T.; Tazi, M.F.; Caution, K.; Hamilton, K.; Badr, A.; Shrestha, C.; Tumin, D.; Hayes, D.; Robledo-Avila, F.; et al. The expression of Mirc1/Mir17–92 cluster in sputum samples correlates with pulmonary exacerbations in cystic fibrosis patients. J. Cyst. Fibros. 2018, 17, 454–461. [Google Scholar] [CrossRef]
- Hassan, T.; McKiernan, P.J.; McElvaney, N.G.; Cryan, S.A.; Greene, C.M. Therapeutic modulation of miRNA for the treatment of proinflammatory lung diseases. Expert Rev. Anti-Infect. Ther. 2012, 10, 359–368. [Google Scholar] [CrossRef]
- Oglesby, I.K.; Vencken, S.F.; Agrawal, R.; Gaughan, K.; Molloy, K.; Higgins, G.; McNally, P.; McElvaney, N.G.; Mall, M.A.; Greene, C.M. miR-17 overexpression in cystic fibrosis airway epithelial cells decreases interleukin-8 production. Eur. Respir. J. 2015, 46, 1350–1360. [Google Scholar] [CrossRef]
- Weldon, S.; McNally, P.; McAuley, D.F.; Oglesby, I.K.; Wohlford-Lenane, C.L.; Bartlett, J.A.; Scott, C.J.; McElvaney, N.G.; Greene, C.M.; McCray, P.B.; et al. miR-31 Dysregulation in Cystic Fibrosis Airways Contributes to Increased Pulmonary Cathepsin S Production. Am. J. Respir. Crit. Care Med. 2014, 190, 165–174. [Google Scholar] [CrossRef]
- Fabbri, E.; Borgatti, M.; Montagner, G.; Bianchi, N.; Finotti, A.; Lampronti, I.; Bezzerri, V.; Dechecchi, M.C.; Cabrini, G.; Gambari, R. Expression of microRNA-93 and Interleukin-8 during Pseudomonas aeruginosa—Mediated Induction of Proinflammatory Responses. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. miR-126 Is Downregulated in Cystic Fibrosis Airway Epithelial Cells and Regulates TOM1 Expression. J. Immunol. 2010, 184, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Cialfi, S.; Cimino, G.; De Biase, R.V.; Dominici, C.; Quattrucci, S.; Pizzuti, A. Elevated levels of miR-145 correlate with SMAD3 down-regulation in Cystic Fibrosis patients. J. Cyst. Fibros. 2013, 12, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Chotirmall, S.H.; McElvaney, N.G.; Greene, C.M. Regulation of Cystic Fibrosis Transmembrane Conductance Regulator by MicroRNA-145, -223, and -494 Is Altered in ΔF508 Cystic Fibrosis Airway Epithelium. J. Immunol. 2013, 190, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Gillen, A.E.; Gosalia, N.; Leir, S.-H.; Harris, A. microRNA regulation of expression of the cystic fibrosis transmembrane conductance regulator gene. Biochem. J. 2011, 438, 25–32. [Google Scholar] [CrossRef]
- Lutful Kabir, F.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-β Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Balakathiresan, N.S.; Dalgard, C.; Gutti, U.; Armistead, D.; Jozwik, C.; Srivastava, M.; Pollard, H.B.; Biswas, R. Elevated miR-155 Promotes Inflammation in Cystic Fibrosis by Driving Hyperexpression of Interleukin-8. J. Biol. Chem. 2011, 286, 11604–11615. [Google Scholar] [CrossRef]
- Zhang, P.-X.; Cheng, J.; Zou, S.; D’Souza, A.D.; Koff, J.L.; Lu, J.; Lee, P.J.; Krause, D.S.; Egan, M.E.; Bruscia, E.M. Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat. Commun. 2015, 6, 6221. [Google Scholar] [CrossRef]
- Ramachandran, S.; Karp, P.H.; Osterhaus, S.R.; Jiang, P.; Wohlford-Lenane, C.; Lennox, K.A.; Jacobi, A.M.; Praekh, K.; Rose, S.D.; Behlke, M.A.; et al. Post-Transcriptional Regulation of Cystic Fibrosis Transmembrane Conductance Regulator Expression and Function by MicroRNAs. Am. J. Respir. Cell Mol. Biol. 2013, 49, 544–551. [Google Scholar] [CrossRef]
- Sonneville, F.; Ruffin, M.; Guillot, L.; Rousselet, N.; Le Rouzic, P.; Corvol, H.; Tabary, O. New Insights about miRNAs in Cystic Fibrosis. Am. J. Pathol. 2015, 185, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, C.; Silswal, N.; Baumlin, N.; Yoshida, M.; Dennis, J.; Yerrathota, S.; Kim, M.; Salathe, M. The CFTR Amplifier Nesolicaftor Rescues TGF-β1 Inhibition of Modulator-Corrected F508del CFTR Function. Int. J. Mol. Sci. 2022, 23, 956. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Model System | CF Population/Genotypes | CFTR Modulator(s) | Key Findings/Correlations | Ref. |
|---|---|---|---|---|
| HNE cells | PwCF homozygous for p.Phe508del or carrying genotypes leading to minimal or residual CFTR function | LUMA TEZA IVA | The level of CFTR rescue significantly correlated with the ppFEV1 change at 6 months in 8 PwCF treated with CFTR modulators. | [25] |
| Ten PwCF (<19 years) with a wide range of CFTR variants | LUMA IVA | CFTR modulation produced changes in CFTR function in nasal and bronchial cell cultures. There was a correlation between the residual CFTR function in both cell types from PwCF with SCC between 60 and 90 mmol·L−1. | [39] | |
| Five healthy subjects (they were not genetically tested) and eight PwCF with variants associated with minimal or residual CFTR function | – | CFTR-mediated transepithelial chloride currents measured in vitro through Ussing chamber assay correlated with the SCC of subjects. | [40] | |
| Twelve adults with CF with either the p.Gly551Asp or p.ArgR117His | IVA | A strong correlation between changes in SCC and in vitro CFTR activation was observed. Furthermore, a moderate correlation between in vitro CFTR activation and changes in ppFEV1 was reported. | [41] | |
| Seven PwCF (≤16 years, clinically stable) with residual CFTR function variants | IVA | All subjects with decreased SCC in response to IVA treatment also had significant increases in chloride current in HNE cultures with IVA exposure. | [42] | |
| Healthy volunteers, PwCF with p.PheF508del/p.Arg117His-7T, p.PheF508del/p.PheF508del, p.PheF508del/p.Met284fs, p.Arg334Trp/c.406-1G>A, and p.Ser18ArgfsX16/p.Ser18ArgfsX16 | LUMA TEZA IVA | In vitro CFTR chloride measurements correlated to changes in SCC. | [43] | |
| Five PwCF: one homozygous for p.Ser737Phe and four compound heterozygous for p.Ser737Phe | TEZA ELX IVA | In vitro analysis demonstrated different levels of CFTR activity. Some degree of CFTR dysfunction was detected by evaluating chloride secretion in HNE cells derived from compound heterozygous subjects; CFTR activity was improved by IVA alone and even more by treatment with correctors. | [44] | |
| Eleven PwCF carrying FDA-eligible variants for ELX/TEZA/IVA and twenty-eight PwCF carrying non-eligible variants | TEZA ELX IVA | There was a significant relationship between CFTR activity correction and changes in ppFEV1 or SCC. | [45] | |
| A 56-year-old male with CF and the p.Phe508del/p.Gln1291His genotype | TEZA ELX IVA | In HNE cells, p.Phe508del/p.Gln1291His resulted in reduced baseline CFTR activity, and showed minimal response to ELX/TEZA/IVA (individually or in combination), aligning with the individual’s clinical evaluation as a non-responder to these drugs. | [46] | |
| Airway organoids | Nine healthy volunteers and three PwCF | LUMA IVA | Organoids treated with modulators displayed similar effects to clinical response; LUMA/IVA treatment promoted greater responses in organoids from p.PheF508del-homozygous subjects. | [26] |
| Six healthy volunteers, nine PwCF homozygous for p.Phe508del, and ten with at least one non-p.Phe508del variant | LUMA IVA | p.Phe508del/p.Phe508del organoids treated with LUMA/IVA exhibited a positive change in swelling responses; a relationship between organoid response to drug and in vivo clinical response was observed for three subjects treated. | [47] | |
| Eighteen PwCF and five healthy volunteers | TEZA ELX IVA | In vitro responses to CFTR modulators correlated well with clinical measurements. | [48] | |
| Five PwCF: p.Phe508del/p.Phe508del, p.Phe508del/p.Gly542X, and F508del/G542X and p.Arg334Trp/p.Arg334Trp | LUMA IVA | Responses of organoids correlated well with clinical findings. | [49] | |
| Intestinal organoids | Twelve healthy volunteers, four CF carriers (WT/p.Phe508del), thirty-five PwCF carrying class II and III variants, and eighteen PwCF carrying class IV and V variants | LUMA IVA | Responses of organoids to CFTR modulators correlated with outcome data from clinical trials. | [24] |
| Twenty-four PwCF: fifteen carrying at least one p.Ser1251Asn and nine carrying at least one (ultra)rare CFTR variant | LUMA IVA | Responses to CFTR modulators in organoids correlated with in vivo measurements (SCC and ppFEV1). | [50] | |
| Thirty-four children with CF carrying a wide range of CFTR variants | – | Organoid swelling significantly correlated with SCC and ICM. | [51] | |
| Thirty-four PwCF with p.Phe508del/p.Phe508del | – | Variability in CFTR residual function appeared to contribute to the clinical heterogeneity and organoid swelling values; responses in organoids correlated with ppFEV1 and BMI. | [52] | |
| Ninety-seven PwCF with well-characterized and rare CFTR variants | LUMA IVA | Measurements of residual CFTR function and rescue by CFTR modulators in organoids correlated with clinical data, namely changes in ppFEV1 and SCC. | [53] | |
| A 56-year-old female with CF carrying p.Phe508del/c.3717+5G>T | LUMA TEZA IVA | No baseline swelling was observed in organoids, suggesting minimal CFTR function; however, limited swelling was detected after LUMA/IVA or TEZA/IVA treatment. | [54] | |
| Twenty-one PwCF homozygous for p.Phe508del | LUMA IVA | No correlations were found between organoid swelling and changes in the in vivo biomarkers, namely SCC and NPD. | [55] | |
| A total of 173 PwCF carrying a wide range of CFTR variants | – | Organoid swelling values were associated with long-term ppFEV1 decline and the probability of developing different CF-related comorbidities, namely pancreatic insufficiency, CF-related liver disease, and CF-related diabetes. | [56] | |
| Fifteen PwCF homozygous for p.Phe508del, fifteen PwCF carrying p.Phe508del/class I variant, and twenty-two PwCF with rare variant non-eligible for CFTR modulator therapy | TEZA ELX IVA | Responses to modulators in organoids from p.Phe508del/p.Phe508del or p.Phe508del/class I variant correlated with changes in ppFEV1. In CF organoids with 11 rare genotypes, CFTR function restoration was reported upon ELX/TEZA/IVA treatment. | [57] | |
| A 19-year-old female with CF carrying p.Tyr515X/p.Arg334Trp | LUMA TEZA ELX IVA | Organoids treated with IVA alone or in combination with correctors demonstrated similar rescue of CFTR-dependent fluid secretion. In vivo measurements demonstrated significant clinical improvements in SCC, ppFEV1, and respiratory symptoms after 7 days of IVA initiation that were sustained for the 9-month follow-up. | [58] | |
| A 6-year-old male with CF carrying p.Phe508del/p.Glu217Gly-Gly509Asp | LUMA TEZA ELX IVA | The p.Glu217Gly-Gly509Asp complex allele was characterized by a high residual function of the CFTR channel; organoid swelling values demonstrated rescue of CFTR function by all tested modulators. | [59] |
| CFTR Modulator(s) | Sample | CF Population/Genotypes | Key Findings | Ref. |
|---|---|---|---|---|
| IVA | Sputum | A total of 151 PwCF (≥6 years) carrying p.Gly551Asp | ↓ bacterial load. | [94] |
| Sputum | Three children with CF carrying p.Gly551Asp | No differences in airway microbiota density. | [95] | |
| Blood | Seven PwCF (≥6 years) carrying at least one residual function variant: p.Gln715X/p.Gly1349Asp; p.Glu1418ArgfsX14/p.Gly1349Asp; p.Gly1349Asp/p.Gly1349Asp; p.Ile1295PhefsX33/p.Gly1349Asp; p.Arg352AlafsX11/p.Gly1349Asp; p.Glu1418ArgfsX14/p.Ser549Arg; p.Phe508del/p.Gly1244Glu | ↑ circulating mononuclear cell count after 2 months of IVA treatment. | [96] | |
| Sputum | Twelve PwCF (≥6 years) carrying p.Gly551Asp | ↓ IL-8, IL-1β and NE levels; ↓ bacterial relative abundance; ↓ arginase-1, myeloperoxidase, calprotectin and S100A9. | [97] | |
| Blood/Sputum | Thirty-three PwCF (≥6 years) carrying p.Gly551Asp | Blood: ↓ CRP, IL-1β, IL-6, IL-8 and IL-10 levels. Sputum: ↓ bacterial relative abundance. | [98] | |
| Sputum | Thirty-one PwCF (≥6 years) carrying p.Gly551Asp | Despite improvements in SCC and ppFEV1, there were no significant changes in airway microbiota density, or in IL-6, IL-8, NE, IL-1β, SLPI, or A1AT levels. | [99] | |
| Epithelial lining fluid | Ten PwCF (≥6 years) carrying p.Gly551Asp | ↓ IL-1β, IL-6, and IL-8 levels. | [100] | |
| Blood | Twenty PwCF carrying p.Gly551Asp | ↓ white blood cell count and IL-6 and IL-8 levels. | [101] | |
| Blood | Ten PwCF (≥6 years) carrying p.Gly551Asp | ↓ CXCL7, IL-8 and sTNFR1 levels. | [102] | |
| BALF | Thirty-nine children with CF carrying at least a gating variant | No differences in neutrophil count and free NE and IL-8 levels. | [103] | |
| LUMA/IVA | Blood | Fourteen PwCF homozygous for p.Phe508del | No differences in white blood cell count or CRP levels. | [104] |
| Blood/Sputum | Fourteen PwCF (≥16 years) homozygous for p.Phe508del | Blood: ↓ IL-8, IL-1β and TNF-α levels. Sputum: ↓ IL-6, IL-8, IL-1β and TNF-α levels. | [105] | |
| Sputum | Thirty PwCF (≥12 years) homozygous for p.Phe508del | ↓ bacterial relative abundance and IL-1β levels. | [106] | |
| Sputum | Forty-one PwCF (≥12 years) homozygous for p.Phe508del | No differences in calprotectin levels or bacterial and fungal relative abundance. | [107] | |
| LUMA/IVA or TEZA/IVA | Blood | LUMA/IVA: 13 PwCF homozygous for p.Phe508del TEZA/IVA: 8 PwCF homozygous for p.Phe508del | LUMA/IVA: ↓ IL-18, TNF-α, and caspase-1 levels. TEZA/IVA: ↓ IL-18, IL-1β, TNF-α, and caspase-1 levels. | [108] |
| ELX/TEZA/IVA | Blood | Forty-eight PwCF carrying at least one p.Phe508del | Of the cultures, 30% were negative to P. aeruginosa, and Meticilin-resistant S. aureus. ↓ IL-6, IL-8, IL-17A levels and neutrophil count. | [109] |
| Sputum | Seventy-nine PwCF (≥12 years) carrying at least one p.Phe508del | ↓ P. aeruginosa relative abundance, IL-1β, IL-8, NE, A1AT, and cathepsin-G levels. ↑ SLPI levels. | [110] | |
| BALF | Eight PwCF (≥12 years) carrying at least one p.Phe508del | ↓ polymorphonuclear cell count. Bacterial cultures were negative for all subjects. | [111] | |
| Blood/Sputum | Thirty PwCF (≥12 years) carrying at least one p.Phe508del | Sputum: ↓ NE, PR3, cathepsin-G, IL-8, IL-1β levels and P. aeruginosa density. ↑ SLPI levels. Blood: ↓ IL-6, CRP, and A1AT levels. | [112] |
| MicroRNA | Target | Key Mechanism(s) | Ref. |
|---|---|---|---|
| miR-17 | IL-8 | ↓ IL-8 levels P. aeruginosa infection reduced miR-17 levels | [143] |
| miR-31 | IRF-1 | ↓ cathepsin-S production | [144] |
| miR-93 | IL-8 | P. aeruginosa infection reduced miR-93 levels and increased IL-8 production | [145] |
| miR-126 | TOM1 | ↑ IL-8 levels and NF-κB signaling | [146] |
| miR-145 | CFTR SMAD3 | Repression of CFTR 3′-UTR reporter ↓ TGF-β signaling | [147,148,149] [147,150] |
| miR-155 | SHIP1 | ↓ SHIP1 levels ↑ IL-8 levels and PI3K/Akt signaling | [151] |
| miR-199a-5p | CAV1 | ↓ CAV1 levels ↑ TLR4 signaling | [152] |
| miR-223 | CFTR | Repression of CFTR 3′-UTR reporter | [148] |
| miR-384 | CFTR and SLC12A2 | ↓ CFTR and SLC12A2 expression levels | [149] |
| miR-494 | CFTR and SLC12A2 | ↓ CFTR and SLC12A2 expression levels Repression of CFTR 3′-UTR reporter ↑ NF-κB signaling, TNF-α, and IL-1β levels increased miR-494 activity | [149] [147,148,149] [153] |
| miR-509-3p | CFTR | ↑ NF-κB signaling, TNF-α, and IL-1β levels increased miR-494 activity | [153] |
| miR-1246 | CFTR and SLC12A2 | ↓ CFTR and SLC12A2 expression levels | [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacalhau, M.; Camargo, M.; Lopes-Pacheco, M. Laboratory Tools to Predict CFTR Modulator Therapy Effectiveness and to Monitor Disease Severity in Cystic Fibrosis. J. Pers. Med. 2024, 14, 93. https://doi.org/10.3390/jpm14010093
Bacalhau M, Camargo M, Lopes-Pacheco M. Laboratory Tools to Predict CFTR Modulator Therapy Effectiveness and to Monitor Disease Severity in Cystic Fibrosis. Journal of Personalized Medicine. 2024; 14(1):93. https://doi.org/10.3390/jpm14010093
Chicago/Turabian StyleBacalhau, Mafalda, Mariana Camargo, and Miquéias Lopes-Pacheco. 2024. "Laboratory Tools to Predict CFTR Modulator Therapy Effectiveness and to Monitor Disease Severity in Cystic Fibrosis" Journal of Personalized Medicine 14, no. 1: 93. https://doi.org/10.3390/jpm14010093
APA StyleBacalhau, M., Camargo, M., & Lopes-Pacheco, M. (2024). Laboratory Tools to Predict CFTR Modulator Therapy Effectiveness and to Monitor Disease Severity in Cystic Fibrosis. Journal of Personalized Medicine, 14(1), 93. https://doi.org/10.3390/jpm14010093