
Clinical and Molecular Characterization of a Patient with Generalized Arterial Calcification of Infancy Caused by Rare ABCC6 Mutation

Abstract
:
1. Introduction
2. Methods
2.1. Whole Exome Sequencing and Copy Number Evaluation
2.2. Real-Time Quantitative PCR
3. Results
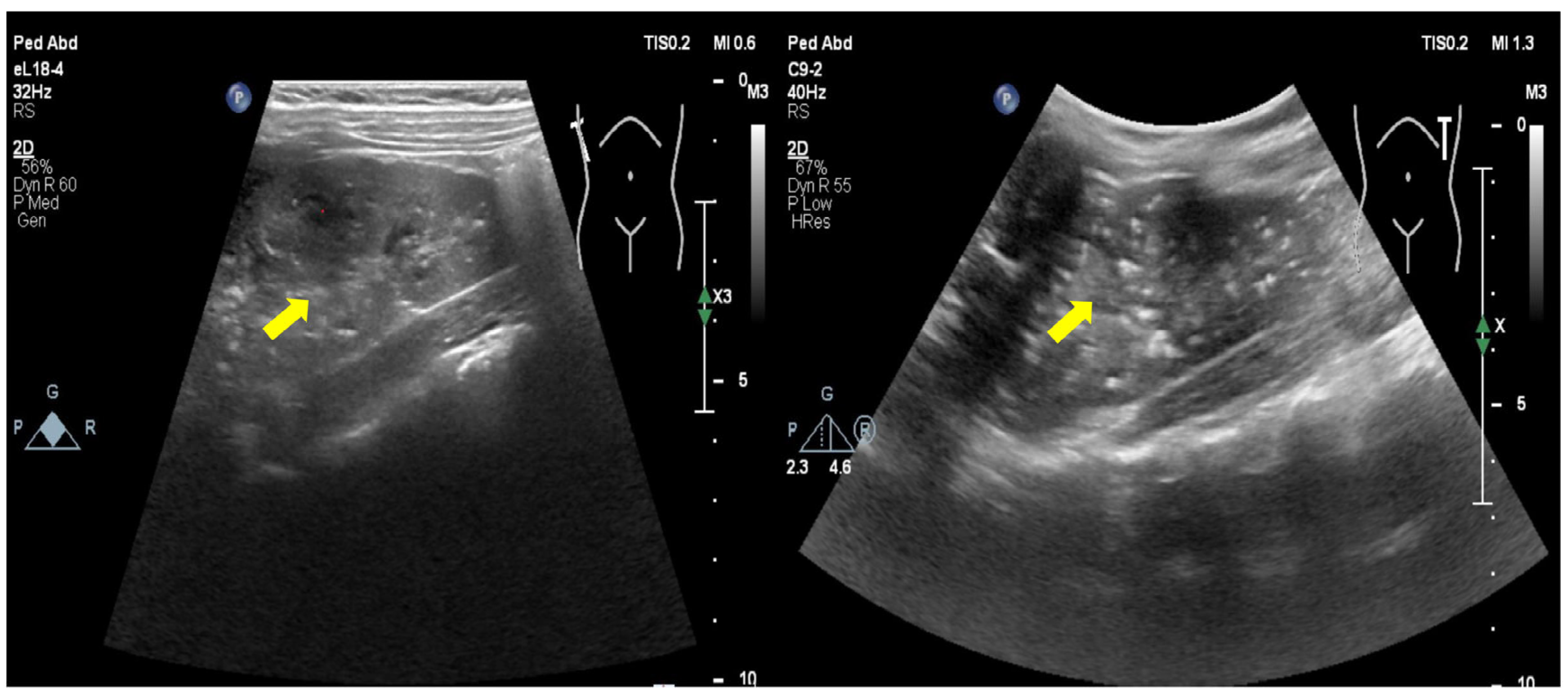
3.1. Clinical Information
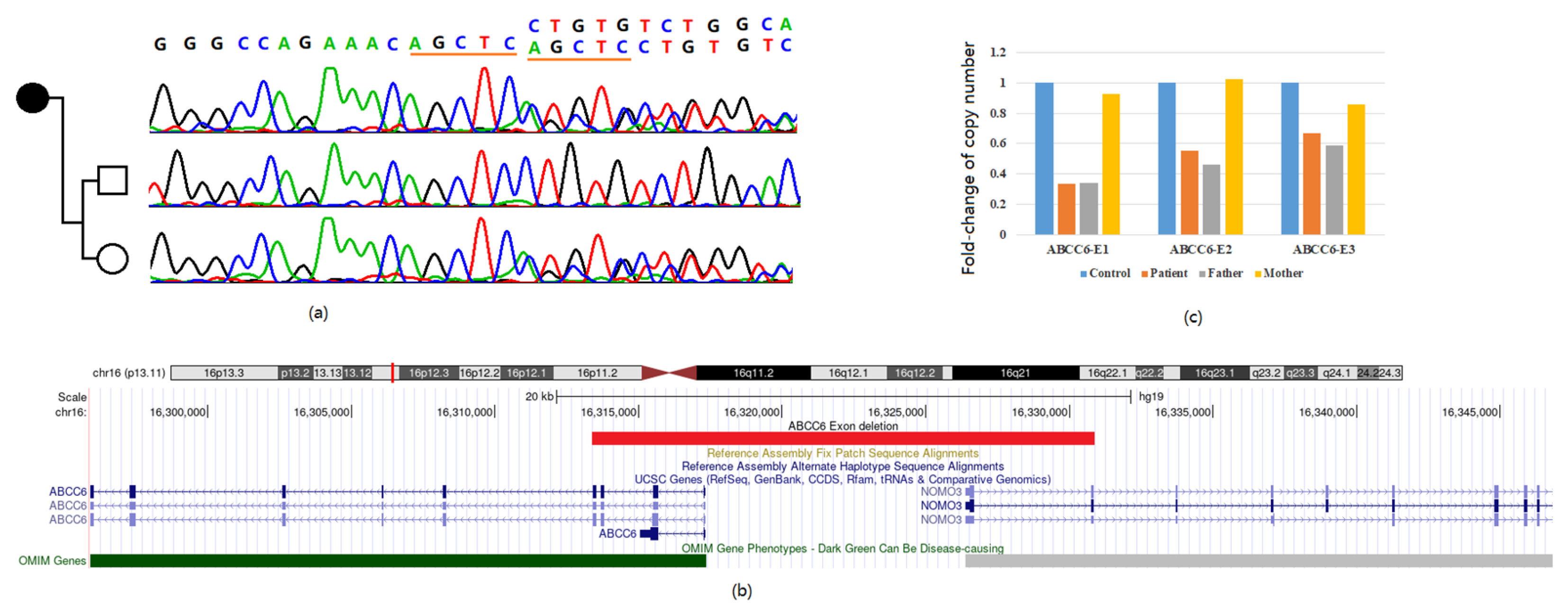
3.2. Genetic Diagnosis Based on Whole Exome Sequencing
3.3. Confirmation of the Exon Deletion
3.4. Reported Cases and Literature Review
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferreira, C.R.; Hackbarth, M.E.; Ziegler, S.G.; Pan, K.S.; Roberts, M.S.; Rosing, D.R.; Whelpley, M.S.; Bryant, J.C.; Macnamara, E.F.; Wang, S.; et al. Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet. Med. 2021, 23, 396–407. [Google Scholar] [CrossRef]
- Choe, Y.; Shin, C.H.; Lee, Y.A.; Kim, M.J.; Lee, Y.J. Case Report and Review of Literature: Autosomal Recessive Hypophosphatemic Rickets Type 2 Caused by a Pathogenic Variant in ENPP1 Gene. Front. Endocrinol. 2022, 13, 911672. [Google Scholar] [CrossRef]
- Akhtar Ali, S.; Ng, C.; Votava-Smith, J.K.; Randolph, L.M.; Pitukcheewanont, P. Bisphosphonate therapy in an infant with generalized arterial calcification with an ABCC6 mutation. Osteoporos. Int. 2018, 29, 2575–2579. [Google Scholar] [CrossRef]
- Bolster, F.; Ali, Z.; Southall, P.; Fowler, D. Generalized arterial calcification of infancy—Findings at post-mortem computed tomography and autopsy. Forensic Sci. Int. 2015, 254, e7–e12. [Google Scholar] [CrossRef]
- Lu, P.; Chen, J.; Chen, M.; Wang, L.; Xiang, D.; Yin, J.; Yang, S. Case report: A rare homozygous variation in the ENPP1 gene, presenting with generalized arterial calcification of infancy in a Chinese infant. Front. Cardiovasc. Med. 2023, 10, 1105381. [Google Scholar] [CrossRef]
- Shimada, B.K.; Pomozi, V.; Zoll, J.; Kuo, S.; Martin, L.; Le Saux, O. ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions. Int. J. Mol. Sci. 2021, 22, 4555. [Google Scholar] [CrossRef]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.F.; et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef]
- Devriese, M.; Legrand, A.; Courtois, M.C.; Jeunemaitre, X.; Albuisson, J. Pseudoxanthoma elasticum with prominent arterial calcifications evoking CD73 deficiency. Vasc. Med. 2019, 24, 461–464. [Google Scholar] [CrossRef]
- Boyce, A.M.; Gafni, R.I.; Ferreira, C.R. Generalized Arterial Calcification of Infancy: New Insights, Controversies, and Approach to Management. Curr. Osteoporos. Rep. 2020, 18, 232–241. [Google Scholar] [CrossRef]
- Li, Q.; van de Wetering, K.; Uitto, J. Pseudoxanthoma Elasticum as a Paradigm of Heritable Ectopic Mineralization Disorders: Pathomechanisms and Treatment Development. Am. J. Pathol. 2019, 189, 216–225. [Google Scholar] [CrossRef]
- Chong, C.R.; Hutchins, G.M. Idiopathic infantile arterial calcification: The spectrum of clinical presentations. Pediatr. Dev. Pathol. 2008, 11, 405–415. [Google Scholar] [CrossRef]
- Kawai, K.; Sato, Y.; Kawakami, R.; Sakamoto, A.; Cornelissen, A.; Mori, M.; Ghosh, S.; Kutys, R.; Virmani, R.; Finn, A.V. Generalized Arterial Calcification of Infancy (GACI): Optimizing Care with a Multidisciplinary Approach. J. Multidiscip. Healthc. 2022, 15, 1261–1276. [Google Scholar] [CrossRef]
- Cheng, Z.; O’Brien, K.; Howe, J.; Sullivan, C.; Schrier, D.; Lynch, A.; Jungles, S.; Sabbagh, Y.; Thompson, D. INZ-701 Prevents Ectopic Tissue Calcification and Restores Bone Architecture and Growth in ENPP1-Deficient Mice. J. Bone Miner. Res. 2021, 36, 1594–1604. [Google Scholar] [CrossRef]
- Jansen, R.S.; Kucukosmanoglu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.; Bergen, A.A.; Gorgels, T.G.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Varadi, A.; Plomp, A.; Bergen, A.A.; Oude Elferink, R.P.; Borst, P.; et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef]
- Albright, R.A.; Stabach, P.; Cao, W.; Kavanagh, D.; Mullen, I.; Braddock, A.A.; Covo, M.S.; Tehan, M.; Yang, G.; Cheng, Z.; et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat. Commun. 2015, 6, 10006. [Google Scholar] [CrossRef]
- Germain, D.P. Pseudoxanthoma elasticum. Orphanet J. Rare Dis. 2017, 12, 85. [Google Scholar] [CrossRef]
- Van Gils, M.; Nollet, L.; Verly, E.; Deianova, N.; Vanakker, O.M. Cellular signaling in pseudoxanthoma elasticum: An update. Cell. Signal. 2019, 55, 119–129. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, Q.; Pfendner, E.; Varadi, A.; Uitto, J. Pseudoxanthoma elasticum: Clinical phenotypes, molecular genetics and putative pathomechanisms. Exp. Dermatol. 2009, 18, 1–11. [Google Scholar] [CrossRef]
- Kozak, E.; Fulop, K.; Tokesi, N.; Rao, N.; Li, Q.; Terry, S.F.; Uitto, J.; Zhang, X.; Becker, C.; Varadi, A.; et al. Oral supplementation of inorganic pyrophosphate in pseudoxanthoma elasticum. Exp. Dermatol. 2022, 31, 548–555. [Google Scholar] [CrossRef]
- Otero, J.E.; Gottesman, G.S.; McAlister, W.H.; Mumm, S.; Madson, K.L.; Kiffer-Moreira, T.; Sheen, C.; Millán, J.L.; Ericson, K.L.; Whyte, M.P. Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2013, 28, 419–430. [Google Scholar] [CrossRef]
- Verschuere, S.; Navassiolava, N.; Martin, L.; Nevalainen, P.I.; Coucke, P.J.; Vanakker, O.M. Reassessment of causality of ABCC6 missense variants associated with pseudoxanthoma elasticum based on Sherloc. Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 131–139. [Google Scholar] [CrossRef]
- Luo, H.; Faghankhani, M.; Cao, Y.; Uitto, J.; Li, Q. Molecular Genetics and Modifier Genes in Pseudoxanthoma Elasticum, a Heritable Multisystem Ectopic Mineralization Disorder. J. Investig. Dermatol. 2021, 141, 1148–1156. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Kintzinger, K.; Hackbarth, M.E.; Botschen, U.; Nitschke, Y.; Mughal, M.Z.; Baujat, G.; Schnabel, D.; Yuen, E.; Gahl, W.A.; et al. Ectopic Calcification and Hypophosphatemic Rickets: Natural History of ENPP1 and ABCC6 Deficiencies. J. Bone Miner. Res. 2021, 36, 2193–2202. [Google Scholar] [CrossRef]
- Nitschke, Y.; Yan, Y.; Buers, I.; Kintziger, K.; Askew, K.; Rutsch, F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Rutsch, F.; Böyer, P.; Nitschke, Y.; Ruf, N.; Lorenz-Depierieux, B.; Wittkampf, T.; Weissen-Plenz, G.; Fischer, R.J.; Mughal, Z.; Gregory, J.W.; et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ. Cardiovasc. Genet. 2008, 1, 133–140. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Role of the extracellular ATP/pyrophosphate metabolism cycle in vascular calcification. Purinergic Signal 2023, 19, 345–352. [Google Scholar] [CrossRef]
- Dedinszki, D.; Szeri, F.; Kozák, E.; Pomozi, V.; Tőkési, N.; Mezei, T.R.; Merczel, K.; Letavernier, E.; Tang, E.; Le Saux, O.; et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol. Med. 2017, 9, 1463–1470. [Google Scholar] [CrossRef]
- Giovannoni, I.; Callea, F.; Travaglini, L.; Amodeo, A.; Cogo, P.; Secinaro, A.; Bizzarri, C.; Cutrera, R.; El Hachem, M.; Francalanci, P. Heart transplant and 2-year follow up in a child with generalized arterial calcification of infancy. Eur. J. Pediatr. 2014, 173, 1735–1740. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence | Product Size |
|---|---|---|
| ABCC6-E1F | 5′ TGCTGGGTCCAAAGTGTTTA 3′ | 469 bp |
| ABCC6-E1R | 5′ CAGCCCGAGAGATCTGCAGC 3′ | |
| ABCC6-E2F | 5′ GATCCAAAAAGTTGCCTGGC 3′ | 328 bp |
| ABCC6-E2R | 5′ TGTCCCCTGCCTCCCCCGAA 3′ | |
| ABCC6-E3F | 5′ CGCCTACCAGTTTGCTGTGA 3′ | 221 bp |
| ABCC6-E3R | 5′ AAGCCGGGCTCCAGACTGAA 3′ | |
| GAPDH-F | 5′ CCCCTTCATACCCTCACGTA 3′ | 192 bp |
| GAPDH-R | 5′ ACACCATCCTAGTTGCCTCC 3′ |
| Parameter | Previous Study | This Study |
|---|---|---|
| ABCC6 Data Set (n = 14) a | ||
| Alive, n (%) | 9 (64.3) | 1 |
| Age at data collection (months), median (range) | 96.9 (2–372) | 60 |
| Deceased, n (%) | 5 (35.7) | 0 |
| Age at death (months), median (range) | 2.6 (1.4–5) | 0 |
| Gender, female/male (% female) | 5/9 (36) | female |
| Mutation type, biallelic/monoallelic (% biallelic) | 8/6 (57.1) | biallelic |
| Rickets | ||
| Yes/no (% yes of assessed) | 1/13 (7.1) | 0 |
| Age at diagnosis (months), median (range) | 2 (2) | 0 |
| Bisphosphonate treatment | ||
| Yes/no (% yes of assessed) | 3/11 (21.4) | 1/0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, R.; Yang, F.; Zhang, Q.; Yu, T.; Yu, Y.; Chang, G.; Wang, X. Clinical and Molecular Characterization of a Patient with Generalized Arterial Calcification of Infancy Caused by Rare ABCC6 Mutation. J. Pers. Med. 2024, 14, 54. https://doi.org/10.3390/jpm14010054
Yao R, Yang F, Zhang Q, Yu T, Yu Y, Chang G, Wang X. Clinical and Molecular Characterization of a Patient with Generalized Arterial Calcification of Infancy Caused by Rare ABCC6 Mutation. Journal of Personalized Medicine. 2024; 14(1):54. https://doi.org/10.3390/jpm14010054
Chicago/Turabian StyleYao, Ruen, Fan Yang, Qianwen Zhang, Tingting Yu, Ying Yu, Guoying Chang, and Xiumin Wang. 2024. "Clinical and Molecular Characterization of a Patient with Generalized Arterial Calcification of Infancy Caused by Rare ABCC6 Mutation" Journal of Personalized Medicine 14, no. 1: 54. https://doi.org/10.3390/jpm14010054
APA StyleYao, R., Yang, F., Zhang, Q., Yu, T., Yu, Y., Chang, G., & Wang, X. (2024). Clinical and Molecular Characterization of a Patient with Generalized Arterial Calcification of Infancy Caused by Rare ABCC6 Mutation. Journal of Personalized Medicine, 14(1), 54. https://doi.org/10.3390/jpm14010054