
Determination of the Duplicated CYP2D6 Allele Using Real-Time PCR Signal: An Alternative Approach

 ,
, 
Abstract
1. Introduction
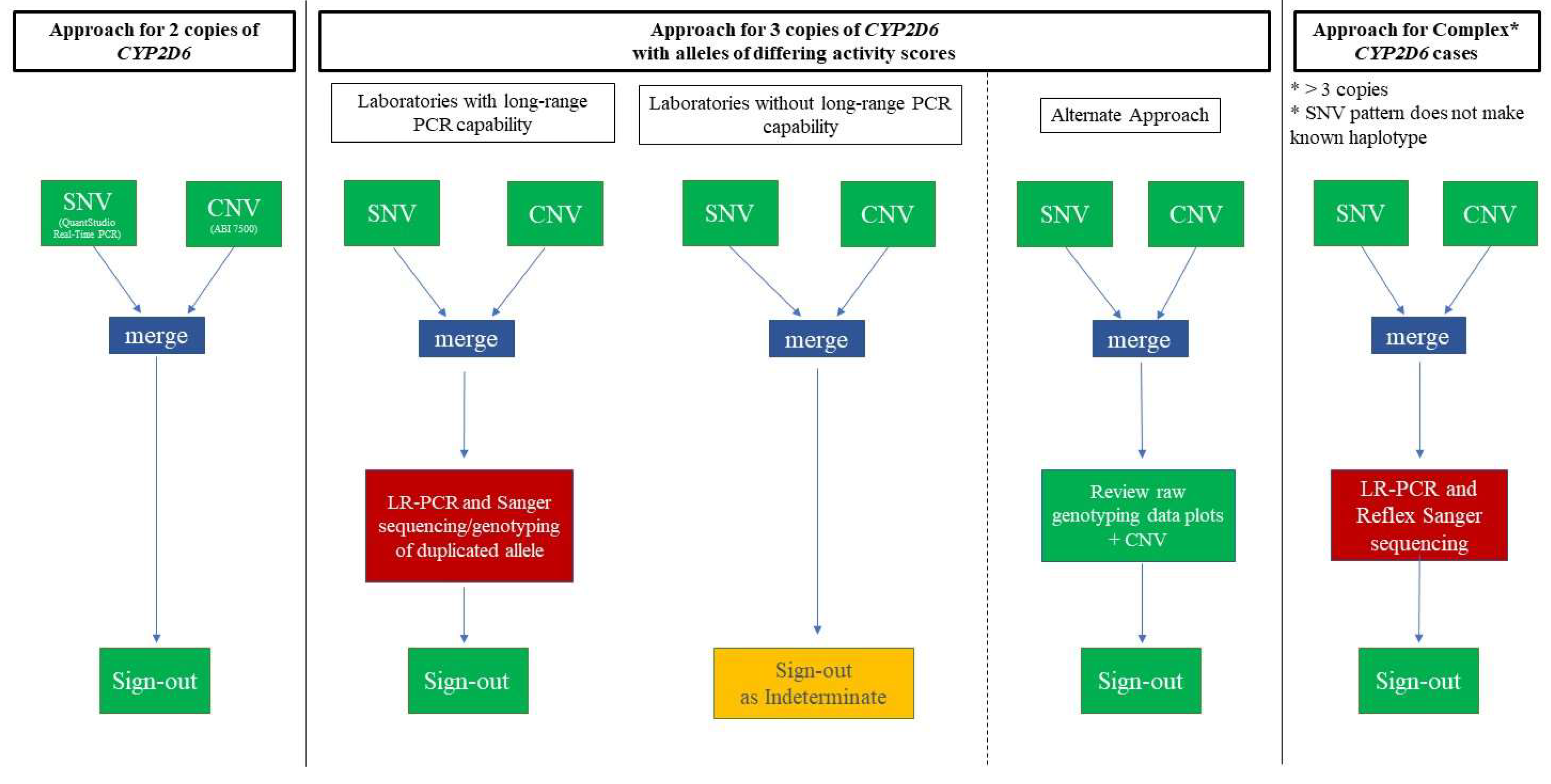
2. Materials and Methods
2.1. Assay Specifications
2.2. CYP2D6-Duplicated-Genotype Determination Exercise
2.3. Statistical Analysis
3. Results
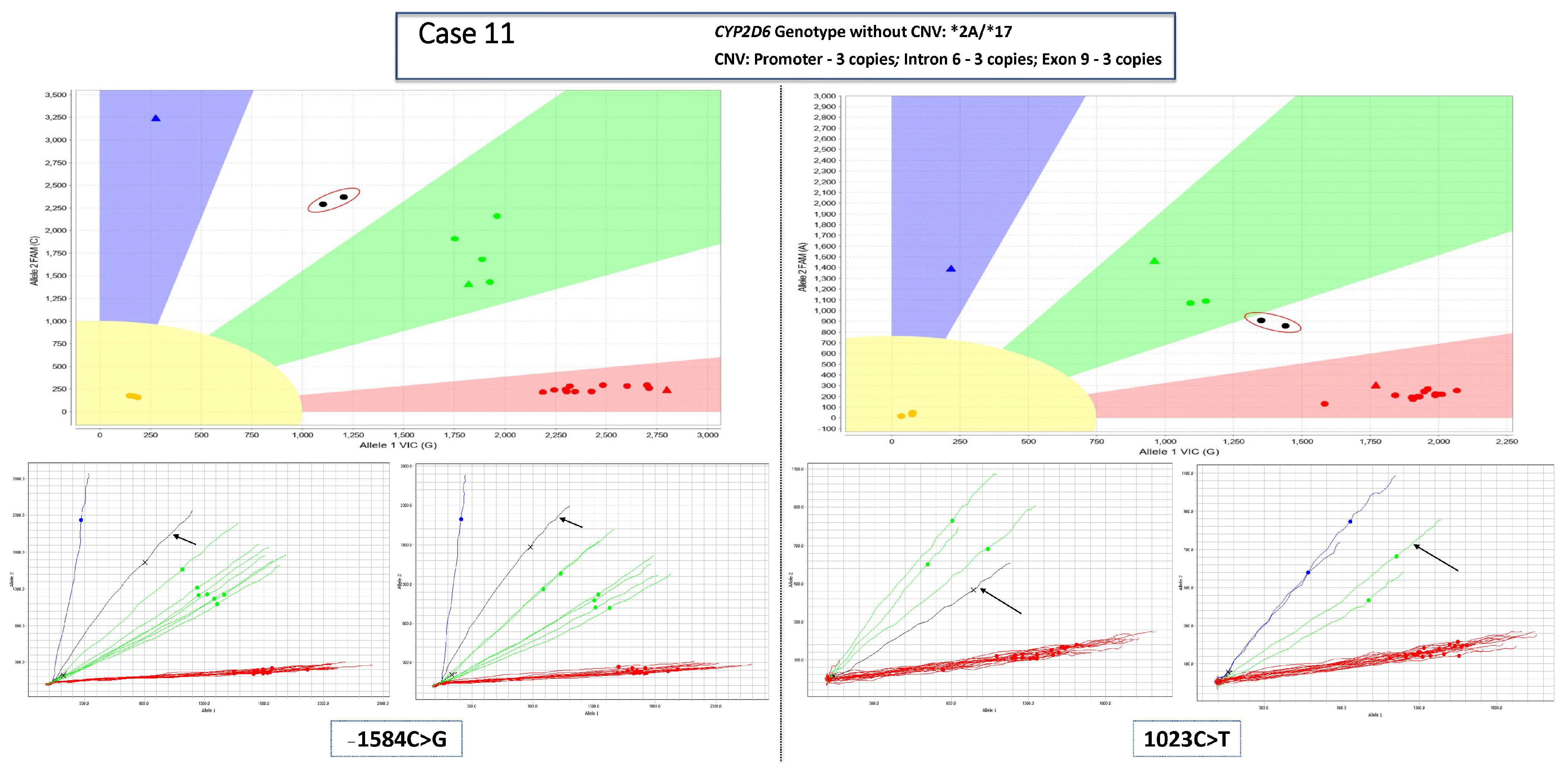
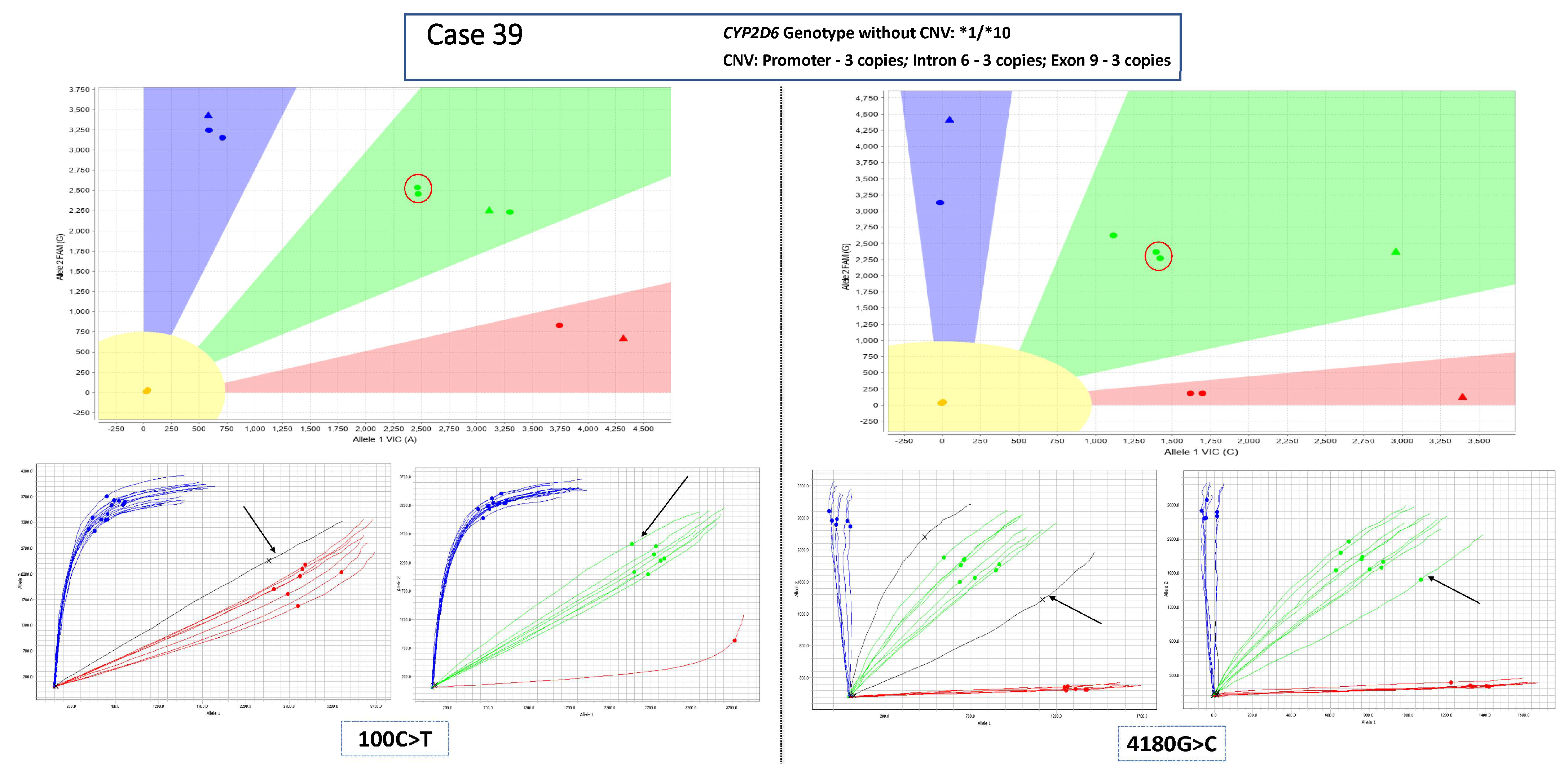
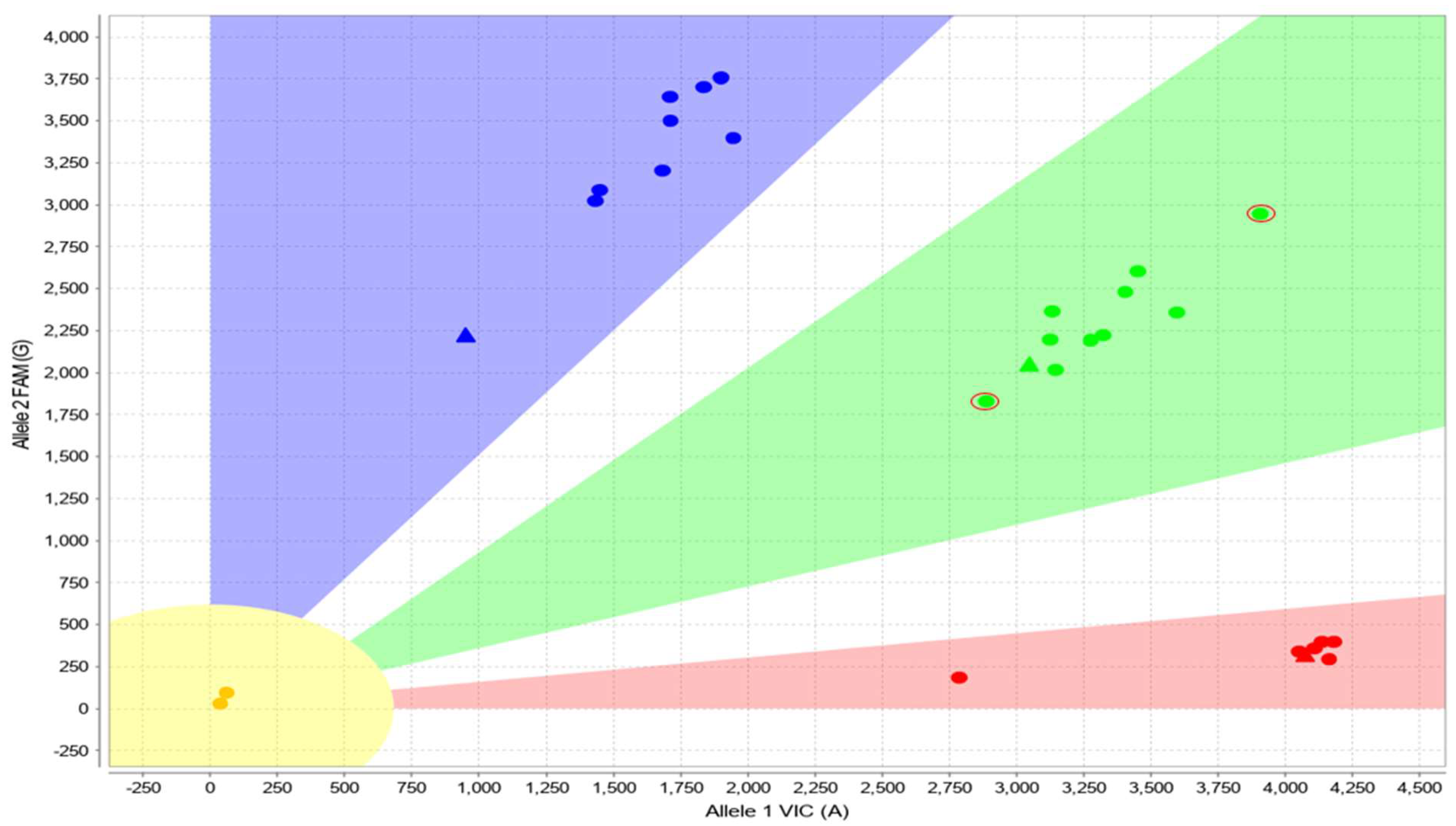
3.1. Main Dataset—Cases with 3 Copies of CYP2D6
3.2. Complex Cases—Including Hybrid Alleles and/or More Than three Copies of CYP2D6
3.3. Cases with Ambiguous CYP2D6 CNV Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esteves, F.; Rueff, J.; Kranendonk, M. The Central Role of Cytochrome P450 in Xenobiotic Metabolism-A Brief Review on a Fascinating Enzyme Family. J. Xenobiot. 2021, 11, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Fuselli, S.; de Filippo, C.; Mona, S.; Sistonen, J.; Fariselli, P.; Destro-Bisol, G.; Barbujani, G.; Bertorelle, G.; Sajantila, A. Evolution of detoxifying systems: The role of environment and population history in shaping genetic diversity at human CYP2D6 locus. Pharm. Genom. 2010, 20, 485–499. [Google Scholar] [CrossRef]
- Hoskins, J.M.; Carey, L.A.; McLeod, H.L. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat. Rev. Cancer 2009, 9, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Penas-Lledo, E.M.; Llerena, A. CYP2D6 variation, behaviour and psychopathology: Implications for pharmacogenomics-guided clinical trials. Br. J. Clin. Pharmacol. 2014, 77, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Crosby, I.; Yip, V.; Maguire, P.; Pirmohamed, M.; Turner, R.M. A Review of the Important Role of CYP2D6 in Pharmacogenomics. Genes 2020, 11, 1295. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef]
- Saravanakumar, A.; Sadighi, A.; Ryu, R.; Akhlaghi, F. Physicochemical Properties, Biotransformation, and Transport Pathways of Established and Newly Approved Medications: A Systematic Review of the Top 200 Most Prescribed Drugs vs. the FDA-Approved Drugs between 2005 and 2016. Clin. Pharm. 2019, 58, 1281–1294. [Google Scholar] [CrossRef]
- CPIC. CYP2D6 CPIC Guidelines. Available online: https://cpicpgx.org/gene/cyp2d6/ (accessed on 13 May 2023).
- Bertilsson, L.; Dahl, M.L.; Dalen, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122. [Google Scholar] [CrossRef]
- Thummler, S.; Dor, E.; David, R.; Leali, G.; Battista, M.; David, A.; Askenazy, F.; Verstuyft, C. Pharmacoresistant Severe Mental Health Disorders in Children and Adolescents: Functional Abnormalities of Cytochrome P450 2D6. Front. Psychiatry 2018, 9, 2. [Google Scholar] [CrossRef]
- Del Tredici, A.L.; Malhotra, A.; Dedek, M.; Espin, F.; Roach, D.; Zhu, G.D.; Voland, J.; Moreno, T.A. Frequency of CYP2D6 Alleles Including Structural Variants in the United States. Front. Pharmacol. 2018, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Nofziger, C.; Paulmichl, M. Accurately genotyping CYP2D6: Not for the faint of heart. Pharmacogenomics 2018, 19, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Ingelman-Sundberg, M.; Miller, N.A.; Leeder, J.S.; Whirl-Carrillo, M.; Klein, T.E.; PharmVar Steering, C. The Pharmacogene Variation (PharmVar) Consortium: Incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin. Pharmacol. Ther. 2018, 103, 399–401. [Google Scholar] [CrossRef]
- Gaedigk, A.; Whirl-Carrillo, M.; Pratt, V.M.; Miller, N.A.; Klein, T.E. PharmVar and the Landscape of Pharmacogenetic Resources. Clin. Pharmacol. Ther. 2020, 107, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Casey, S.T.; Whirl-Carrillo, M.; Miller, N.A.; Klein, T.E. Pharmacogene Variation Consortium: A Global Resource and Repository for Pharmacogene Variation. Clin. Pharmacol. Ther. 2021, 110, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Pharmacogene Variation Consortium. CYP2D6. Available online: https://www.pharmvar.org/gene/CYP2D6 (accessed on 13 May 2023).
- Jarvis, J.P.; Peter, A.P.; Shaman, J.A. Consequences of CYP2D6 Copy-Number Variation for Pharmacogenomics in Psychiatry. Front. Psychiatry 2019, 10, 432. [Google Scholar] [CrossRef] [PubMed]
- Beoris, M.; Amos Wilson, J.; Garces, J.A.; Lukowiak, A.A. CYP2D6 copy number distribution in the US population. Pharm. Genom. 2016, 26, 96–99. [Google Scholar] [CrossRef][Green Version]
- Pratt, V.M.; Cavallari, L.H.; Del Tredici, A.L.; Gaedigk, A.; Hachad, H.; Ji, Y.; Kalman, L.V.; Ly, R.C.; Moyer, A.M.; Scott, S.A.; et al. Recommendations for Clinical CYP2D6 Genotyping Allele Selection: A Joint Consensus Recommendation of the Association for Molecular Pathology, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, and the European Society for Pharmacogenomics and Personalized Therapy. J. Mol. Diagn. 2021, 23, 1047–1064. [Google Scholar] [CrossRef]
- Black, J.L., 3rd; Walker, D.L.; O'Kane, D.J.; Harmandayan, M. Frequency of undetected CYP2D6 hybrid genes in clinical samples: Impact on phenotype prediction. Drug Metab. Dispos. 2012, 40, 111–119. [Google Scholar] [CrossRef]
- Cavallari, L.H.; Van Driest, S.L.; Prows, C.A.; Bishop, J.R.; Limdi, N.A.; Pratt, V.M.; Ramsey, L.B.; Smith, D.M.; Tuteja, S.; Duong, B.Q.; et al. Multi-site investigation of strategies for the clinical implementation of CYP2D6 genotyping to guide drug prescribing. Genet. Med. 2019, 21, 2255–2263. [Google Scholar] [CrossRef]
- Moyer, A.M.; McMillin, G.A.; Long, T.A.; Gandhi, M.J.; Mao, R.; Smock, K.J.; Halley, J.G.; Weck, K.E. Genotype and Phenotype Concordance for Pharmacogenetic Tests through Proficiency Survey Testing. Arch. Pathol. Lab. Med. 2020, 144, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Carvalho Henriques, B.; Buchner, A.; Hu, X.; Wang, Y.; Yavorskyy, V.; Wallace, K.; Dong, R.; Martens, K.; Carr, M.S.; Behroozi Asl, B.; et al. Methodology for clinical genotyping of CYP2D6 and CYP2C19. Transl. Psychiatry 2021, 11, 596. [Google Scholar] [CrossRef] [PubMed]
- Kramer, W.E.; Walker, D.L.; O'Kane, D.J.; Mrazek, D.A.; Fisher, P.K.; Dukek, B.A.; Bruflat, J.K.; Black, J.L. CYP2D6: Novel genomic structures and alleles. Pharm. Genom. 2009, 19, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, K.A. Computing Inter-Rater Reliability for Observational Data: An Overview and Tutorial. Tutor. Quant. Methods Psychol. 2012, 8, 23–34. [Google Scholar] [CrossRef]
- McHugh, M.L. Interrater reliability: The kappa statistic. Biochem. Med. 2012, 22, 276–282. [Google Scholar] [CrossRef]
- Sicko, R.J.; Romitti, P.A.; Browne, M.L.; Brody, L.C.; Stevens, C.F.; Mills, J.L.; Caggana, M.; Kay, D.M. Rare Variants in RPPH1 Real-Time Quantitative PCR Control Assay Binding Sites Result in Incorrect Copy Number Calls. J. Mol. Diagn. 2022, 24, 33–40. [Google Scholar] [CrossRef]
- Larsen, J.B.; Jorgensen, S. Simple and Robust Detection of CYP2D6 Gene Deletions and Duplications Using CYP2D8P as Reference. Pharmaceuticals 2022, 15, 166. [Google Scholar] [CrossRef]
- Riffel, A.K.; Dehghani, M.; Hartshorne, T.; Floyd, K.C.; Leeder, J.S.; Rosenblatt, K.P.; Gaedigk, A. CYP2D7 Sequence Variation Interferes with TaqMan CYP2D6 (*) 15 and (*) 35 Genotyping. Front. Pharmacol. 2015, 6, 312. [Google Scholar] [CrossRef]
- Gaedigk, A.; Riffel, A.K.; Leeder, J.S. CYP2D6 Haplotype Determination Using Long Range Allele-Specific Amplification: Resolution of a Complex Genotype and a Discordant Genotype Involving the CYP2D6*59 Allele. J. Mol. Diagn. 2015, 17, 740–748. [Google Scholar] [CrossRef]
- Rasmussen, H.B.; Werge, T. Novel variant of CYP2D6*6 is undetected by a commonly used genotyping procedure. Pharmacol. Rep. 2011, 63, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.J.; Aggarwal, P.; Boone, E.C.; Haidar, C.E.; Relling, M.V.; Derezinski, A.D.; Broeckel, U.; Gaedigk, A. Identification of CYP2D6 Haplotypes that Interfere with Commonly Used Assays for Copy Number Variation Characterization. J. Mol. Diagn. 2021, 23, 577–588. [Google Scholar] [CrossRef]
- Scantamburlo, G.; Tziolia, K.; Zopf, M.; Bernardinelli, E.; Soyal, S.M.; Civello, D.A.; Vanoni, S.; Dossena, S.; Patsch, W.; Patrinos, G.P.; et al. Allele Drop out Conferred by a Frequent CYP2D6 Genetic Variation For Commonly Used CYP2D6*3 Genotyping Assays. Cell Physiol. Biochem. 2017, 43, 2297–2309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tremmel, R.; Schaeffeler, E.; Schwab, M.; Lauschke, V.M. Challenges and opportunities associated with rare-variant pharmacogenomics. Trends Pharmacol. Sci. 2022, 43, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Scherer, S.E.; Bielinski, S.J.; Muzny, D.M.; Jones, L.A.; Black, J.L., 3rd; Moyer, A.M.; Giri, J.; Sharp, R.R.; Matey, E.T.; et al. Implementation of preemptive DNA sequence-based pharmacogenomics testing across a large academic medical center: The Mayo-Baylor RIGHT 10K Study. Genet. Med. 2022, 24, 1062–1072. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 26. [Google Scholar] [CrossRef]
- Yang, Y.; Botton, M.R.; Scott, E.R.; Scott, S.A. Sequencing the CYP2D6 gene: From variant allele discovery to clinical pharmacogenetic testing. Pharmacogenomics 2017, 18, 673–685. [Google Scholar] [CrossRef]
- Liau, Y.; Maggo, S.; Miller, A.L.; Pearson, J.F.; Kennedy, M.A.; Cree, S.L. Nanopore sequencing of the pharmacogene CYP2D6 allows simultaneous haplotyping and detection of duplications. Pharmacogenomics 2019, 20, 1033–1047. [Google Scholar] [CrossRef]
- Twist, G.P.; Gaedigk, A.; Miller, N.A.; Farrow, E.G.; Willig, L.K.; Dinwiddie, D.L.; Petrikin, J.E.; Soden, S.E.; Herd, S.; Gibson, M.; et al. Erratum: Constellation: A tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. NPJ Genom. Med. 2017, 2, 16039. [Google Scholar] [CrossRef]
- Lee, S.B.; Wheeler, M.M.; Patterson, K.; McGee, S.; Dalton, R.; Woodahl, E.L.; Gaedigk, A.; Thummel, K.E.; Nickerson, D.A. Stargazer: A software tool for calling star alleles from next-generation sequencing data using CYP2D6 as a model. Genet. Med. 2019, 21, 361–372. [Google Scholar] [CrossRef]
- Qiao, W.; Wang, J.; Pullman, B.S.; Chen, R.; Yang, Y.; Scott, S.A. The CYP2D6 VCF Translator. Pharm. J. 2017, 17, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Numanagic, I.; Malikic, S.; Ford, M.; Qin, X.; Toji, L.; Radovich, M.; Skaar, T.C.; Pratt, V.M.; Berger, B.; Scherer, S.; et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat. Commun. 2018, 9, 828. [Google Scholar] [CrossRef] [PubMed]
- Twesigomwe, D.; Drogemoller, B.I.; Wright, G.E.B.; Siddiqui, A.; da Rocha, J.; Lombard, Z.; Hazelhurst, S. StellarPGx: A Nextflow Pipeline for Calling Star Alleles in Cytochrome P450 Genes. Clin. Pharmacol. Ther. 2021, 110, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, F.; Gonzaludo, N.; Malhotra, A.; Rogert, C.; Taft, R.J.; Bentley, D.R.; Eberle, M.A. Cyrius: Accurate CYP2D6 genotyping using whole-genome sequencing data. Pharm. J. 2021, 21, 251–261. [Google Scholar] [CrossRef]
- Twesigomwe, D.; Wright, G.E.B.; Drogemoller, B.I.; da Rocha, J.; Lombard, Z.; Hazelhurst, S. A systematic comparison of pharmacogene star allele calling bioinformatics algorithms: A focus on CYP2D6 genotyping. NPJ Genom. Med. 2020, 5, 30. [Google Scholar] [CrossRef]
- Hari, A.; Zhou, Q.; Gonzaludo, N.; Harting, J.; Scott, S.A.; Qin, X.; Scherer, S.; Sahinalp, S.C.; Numanagic, I. An efficient genotyper and star-allele caller for pharmacogenomics. Genome Res. 2023, 33, 61–70. [Google Scholar] [CrossRef]
- Cohn, I.; Paton, T.A.; Marshall, C.R.; Basran, R.; Stavropoulos, D.J.; Ray, P.N.; Monfared, N.; Hayeems, R.Z.; Meyn, M.S.; Bowdin, S.; et al. Genome sequencing as a platform for pharmacogenetic genotyping: A pediatric cohort study. NPJ Genom. Med. 2017, 2, 19. [Google Scholar] [CrossRef]
- Lopes, J.L.; Harris, K.; Karow, M.B.; Peterson, S.E.; Kluge, M.L.; Kotzer, K.E.; Lopes, G.S.; Larson, N.B.; Bielinski, S.J.; Scherer, S.E.; et al. Targeted Genotyping in Clinical Pharmacogenomics: What Is Missing? J. Mol. Diagn. 2022, 24, 253–261. [Google Scholar] [CrossRef]
- Ammar, R.; Paton, T.A.; Torti, D.; Shlien, A.; Bader, G.D. Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Res 2015, 4, 17. [Google Scholar] [CrossRef]
- Qiao, W.; Yang, Y.; Sebra, R.; Mendiratta, G.; Gaedigk, A.; Desnick, R.J.; Scott, S.A. Long-Read Single Molecule Real-Time Full Gene Sequencing of Cytochrome P450-2D6. Hum. Mutat. 2016, 37, 315–323. [Google Scholar] [CrossRef]
- Van der Lee, M.; Allard, W.G.; Vossen, R.; Baak-Pablo, R.F.; Menafra, R.; Deiman, B.; Deenen, M.J.; Neven, P.; Johansson, I.; Gastaldello, S.; et al. Toward predicting CYP2D6-mediated variable drug response from CYP2D6 gene sequencing data. Sci. Transl. Med. 2021, 13, eabf3637. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Dataset: Cases with 3 Copies of CYP2D6 (n = 73) | ||||||
|---|---|---|---|---|---|---|
| Genotype | Total Cases | Phenotype | Activity Score | Number of Het SNVs | Number of Reviewers Who Requested Sequencing for Each Case † | |
| *1/*4×2 | 11 | Intermediate | 1 | 3 | 0 | |
| 0 | ||||||
| 0 | ||||||
| 0 | ||||||
| 2 | ||||||
| 0 | ||||||
| 1 | ||||||
| 0 | ||||||
| 0 | ||||||
| 3 | ||||||
| 1 | ||||||
| *1×2/*4 | 7 | Normal | 2 | 3 | 1 | |
| 0 | ||||||
| 0 | ||||||
| 0 | ||||||
| 1 | ||||||
| 2 | ||||||
| 0 | ||||||
| *1×2/*41 | 6 | Ultrarapid | 2.5 | 3 | 1 | |
| 1 | ||||||
| 0 | ||||||
| 1 | ||||||
| 1 | ||||||
| 1 | ||||||
| *1/*2×2 | 6 | Ultrarapid | 3 | 2 | 0 | |
| 0 | ||||||
| 0 | ||||||
| 0 | ||||||
| 0 | ||||||
| 0 | ||||||
| *4×2/*17 | 5 | Intermediate | 0.5 | 4 | 0 | |
| 1 | ||||||
| 1 | ||||||
| 0 | ||||||
| 0 | ||||||
| *2A×2/*41 | 4 | Ultrarapid | 2.5 | 2 | 1 | |
| 2 | ||||||
| 1 | ||||||
| 4 | ||||||
| *1×2/*9 | 4 | Ultrarapid | 2.5 | 1 | 1 | |
| 1 | ||||||
| 1 | ||||||
| 1 | ||||||
| *2A×2/*4 | 3 | Normal | 2 | 4 | 0 | |
| 0 | ||||||
| 0 | ||||||
| *2A×2/*17 | 2 | Ultrarapid | 2.5 | 2 | 0 | |
| 0 | ||||||
| *1/*41×2 | 2 | Normal | 2 | 3 | 1 | |
| 0 | ||||||
| *2×2/*9 | 2 | Ultrarapid | 2.5 | 3 | 5 | |
| 3 | ||||||
| *2×2/*2A | 2 | Ultrarapid | 3 | 1 | 2 | |
| 2 | ||||||
| *1×2/*10 | 2 | Normal | 2.25 | 2 | 2 | |
| 6 | ||||||
| *4/*35×2 | 1 | Normal | 2 | 3 | 0 | |
| *27×2/*41 | 1 | Ultrarapid | 2.5 | 2 | 2 | |
| *9/*35×2 | 1 | Ultrarapid | 2.5 | 5 | 1 | |
| *4×2/*59 | 1 | Intermediate | 0.5 | 4 | 3 | |
| *4×2/*41 | 1 | Intermediate | 0.5 | 4 | 1 | |
| *4×2/*35 | 1 | Intermediate | 1 | 5 | 1 | |
| *2×2/*35 | 1 | Ultrarapid | 3 | 2 | 2 | |
| *2×2/*4 | 1 | Normal | 2 | 1 | 1 | |
| *2A×2/*29 | 1 | Ultrarapid | 2.5 | 2 | 2 | |
| *2A×2/*10 | 1 | Normal | 2.25 | 3 | 0 | |
| *2A×2/*9 | 1 | Ultrarapid | 2.5 | 4 | 0 | |
| *2A×2/*6 | 1 | Normal | 2 | 4 | 0 | |
| *2A/*4×2 | 1 | Intermediate | 1 | 4 | 0 | |
| *2/*4×2 | 1 | Intermediate | 1 | 3 | 1 | |
| *1×2/*6 | 1 | Normal | 2 | 1 | 1 | |
| *1×2/*3 | 1 | Normal | 2 | 1 | 1 | |
| *1/*2A×2 | 1 | Ultrarapid | 3 | 3 | 0 | |
| Exploratory Data Set: Cases with Hybrid Alleles and/or >3 copies of CYP2D6 (n = 11) | ||||||
| Genotype | CNV § | Total Cases | Phenotype | Activity Score | Number of Het SNVs | Number of Reviewers Requesting Sequencing † |
| *68+4/*41×2 | 4, 3, 3 | 2 | Intermediate | 1 | 4 | 1/4 |
| 2/4 | ||||||
| *1×2/*68+*4 | 4, 3, 3 | 2 | Normal | 2 | 3 | 1/4 |
| 2/3 | ||||||
| *2A×2/*68+*4 | 4, 3, 3 | 1 | Normal | 2 | 4 | 1/4 |
| *1×N/*68+*4 | 6, 6, 5 | 1 | Ultrarapid | >3 | 3 | 4/4 |
| *13+*2A×2/*9 | 3, 4, 4 | 1 | Ultrarapid | 2.5 | 4 | 1/4 |
| *1×2/*36+*10 | 4, 4, 3 | 1 | Normal | 2 | 2 | 4/4 |
| *4×2/*29×2 | 4, 4, 4 | 1 | Intermediate | 0.5 | 4 | 4/4 |
| *2A×2/*2+*17 or *2A×2+*2/*17 | 4, 4, 4 | 1 | Ultrarapid | 3.5 | 2 | 4/4 |
| *2A×2/*4 | 14, 3, 3 | 1 | Normal | 2 | 4 | 4/4 |
| Ambiguous CNV Status between 2 and 3 (n = 3) | ||||||
| Genotype | Total Cases | Phenotype | Activity Score | Number of Het SNVs | ||
| *1/*6 | 1 | Intermediate | 1 | 1 | ||
| *2A/*35 | 1 | Normal | 2 | 1 | ||
| *9/*35 | 1 | Normal | 1.5 | 5 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atiq, M.A.; Peterson, S.E.; Langman, L.J.; Baudhuin, L.M.; Black, J.L.; Moyer, A.M. Determination of the Duplicated CYP2D6 Allele Using Real-Time PCR Signal: An Alternative Approach. J. Pers. Med. 2023, 13, 883. https://doi.org/10.3390/jpm13060883
Atiq MA, Peterson SE, Langman LJ, Baudhuin LM, Black JL, Moyer AM. Determination of the Duplicated CYP2D6 Allele Using Real-Time PCR Signal: An Alternative Approach. Journal of Personalized Medicine. 2023; 13(6):883. https://doi.org/10.3390/jpm13060883
Chicago/Turabian StyleAtiq, Mazen A., Sandra E. Peterson, Loralie J. Langman, Linnea M. Baudhuin, John L. Black, and Ann M. Moyer. 2023. "Determination of the Duplicated CYP2D6 Allele Using Real-Time PCR Signal: An Alternative Approach" Journal of Personalized Medicine 13, no. 6: 883. https://doi.org/10.3390/jpm13060883
APA StyleAtiq, M. A., Peterson, S. E., Langman, L. J., Baudhuin, L. M., Black, J. L., & Moyer, A. M. (2023). Determination of the Duplicated CYP2D6 Allele Using Real-Time PCR Signal: An Alternative Approach. Journal of Personalized Medicine, 13(6), 883. https://doi.org/10.3390/jpm13060883