
Real-World Evaluation of a Population Germline Genetic Screening Initiative for Family Medicine Patients

 ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Setting
2.2. Patient Selection
2.3. Germline Sequencing, Pathogenic Variant Interpretation and Reporting of Results
2.4. Patient Follow-Up and Clinical Impact
2.5. Determination of Relevance to Therapy or Disease
2.6. Statistical Analysis
3. Results
3.1. Study Population
3.2. Genomic Variant Frequencies
3.3. Clinical Impact: Pharmacogenes
3.4. Clinical Impact: Hereditary Predisposition Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Gene | Detectable Variants |
|---|---|
| CACNA1S | All pathogenic/likely pathogenic variants a |
| CFTR | *c.1652G>A |
| CYP2C19 | *7 |
| CYP2C9 | *3, *5, *6 |
| CYP2D6 | *50 |
| CYP3A5 | *6, *7 |
| CYP4F2 | *3 |
| DPYD | *2A, *HapB3, *3, *7, *8, *10, *12, *13, *c.557A>G, *c.2846A>T |
| G6PD | Class I deficiency |
| G6PD | Class II deficiency |
| G6PD | Class III deficiency |
| HLA-B | *57:01, *15:02, *58:01 b |
| RYR1 | All pathogenic/likely pathogenic variants a |
| SLCO1B1 | *5, *15/*17 c |
| TPMT | *2, *3A, *3B, *3C, *4, *11, *14, *15, *23, *29, *41 |
| VKORC1 | *1173C>T (in linkage with c.-1639G>A) |
| Gene | Phenotype |
|---|---|
| BRCA1 BRCA2 | Hereditary breast and ovarian cancer |
| TP53 | Li Fraumeni syndrome |
| STK11 | Peutz-Jeghers syndrome |
| MLH1 MSH2 MSH6 PMS2 | Lynch syndrome |
| APC | Familial adenomatous polyposis |
| MUTYH | MYH-associated polyposis |
| BMPR1A SMAD4 | Juvenile polyposis |
| VHL | Von Hippel–Lindau syndrome |
| MEN1 | Multiple endocrine neoplasia type 1 |
| RET | Multiple endocrine neoplasia type 2 Familial medullary thyroid cancer |
| PTEN | PTEN hamartoma tumor syndrome |
| RB1 | Retinoblastoma |
| SDHD SDHAF2 SDHC SDHB | Hereditary paraganglioma-pheochromocytoma syndrome |
| TSC1 TSC2 | Tuberous sclerosis |
| WT1 | WT1-related Wilms tumor |
| NF2 | Neurofibromatosis type 2 |
| COL3A1 | Ehlers–Danlos syndrome, vascular type |
| FBN1 | Marfan syndrome |
| TGFBR1 TGFBR2 SMAD3 | Loeys–Dietz syndrome |
| ACTA2 MYH11 | Familial thoracic aortic aneurysms and dissections |
| MYBPC3 MYH7 TNNT2 TNNI3 TPM1 MYL3 ACTC1 PRKAG2 GLA MYL2 LMNA | Hypertrophic and dilated cardiomyopathy |
| RYR2 | Catecholaminergic polymorphic ventricular tachycardia |
| PKP2 DSP DSC2 TMEM43 DSG2 | Arrhythmogenic right ventricular cardiomyopathy |
| KCNQ1 KCNH2 SCN5A | Romano-Ward long QT syndromes (types 1, 2, and 3), Brugada syndrome |
| LDLR APOB PCSK9 | Familial hypercholesterolemia |
| ATP7B | Wilson’s disease |
| OTC | Ornithine transcarbamylase deficiency |
| RYR1 CACNA1S | Malignant hyperthermia susceptibility |
| Pharmacogene | Drug(s) |
|---|---|
| CACNA1S | Desflurane Enflurane Halothane Isoflurane Methoxyflurane Sevoflurane Succinylcholine |
| CFTR | Ivacaftor |
| CYP2B6 a | Efavirenz |
| CYP2C19 | Amitriptyline Citalopram Clopidogrel Escitalopram Lansoprazole Omeprazole Pantoprazole Voriconazole |
| CYP2C9 | Celecoxib Flubiprofen Fosphenytoin Ibuprofen Lornoxicam Meloxicam Phenytoin Piroxicam Siponimod Tenoxicam Warfarin |
| CYP2D6 | Amitriptyline Atomoxetine Codeine Nortriptyline Ondansetron Paroxetine Pitolisant Tamoxifen Tramadol Tropisetron |
| CYP3A5 | Tamoxifen |
| CYP4F2 | Warfarin |
| DPYD | Capecitabine Fluorouracil |
| G6PD | Rasburicase Tafenoquine |
| HLA-Aa | Carbamazepine |
| HLA-B | Abacavir Allopurinol Carbamazepine Fosphenytoin Oxcarbazepine Phenytoin |
| IFNL3a | Peginterferon Alfa-2a Peginterferon Alfa-2b |
| IFNL4a | Peginterferon Alfa-2a Peginterferon Alfa-2b |
| NUDT15a | Azathioprine Mercaptopurine Thioguanine |
| RYR1 | Desflurane Enflurane Halothane Isoflurane Methoxyflurane Sevoflurane Succinylcholine |
| SLCO1B1 | Simvastatin |
| TPMT | Azathioprine Mercaptopurine Thioguanine |
| UGT1A1a | Atazanavir Irinotecan |
| VKORC1 | Warfarin |
| Carrier Frequency | |||||
|---|---|---|---|---|---|
| ACMG SF a Gene Mutation | UKFM (n = 205) | Geisinger (n = 1415) b | p-Value | OR (95% CI) | Adjusted p-Value c |
| Any d | 10 (4.9%) | 46 (3.3%) | 0.222 | 1.53 (0.68, 3.13) | N/A |
| MUTYHd | 3 (1.5%) | N/A | N/A | N/A | N/A |
| CACNA1S | 2 (1.0%) | 0 (0.0%) | 0.016 | Inf (1.30, Inf) | 0.144 |
| KCNQ1 | 2 (1.0%) | 0 (0.0%) | 0.016 | Inf (1.30, Inf) | 0.144 |
| APOB | 1 (0.5%) | 2 (0.1%) | 0.334 | 3.46 (0.06, 66.82) | 1.000 |
| BRCA2 | 1 (0.5%) | 6 (0.4%) | 1.000 | 1.15 (0.03, 9.56) | 1.000 |
| DSC2 | 1 (0.5%) | 0 (0.0%) | 0.127 | Inf (0.18, Inf) | 0.854 |
| LDLR | 1 (0.5%) | 5 (0.4%) | 0.557 | 1.38 (0.03, 12.44) | 1.000 |
| SCN5A | 1 (0.5%) | 3 (0.2%) | 0.418 | 2.31 (0.04, 28.87) | 1.000 |
| SDHB | 1 (0.5%) | 1 (0.1%) | 0.237 | 6.93 (0.09, 541.92) | 1.000 |
References
- Gupta, S.; Weiss, J.M.; Axell, L.; Burke, C.A.; Chen, L.-M.; Chung, D.C.; Clayback, K.M.; Dallas, S.; Felder, S.; Giardiello, F.M.; et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Colorectal. Version 2. 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (accessed on 26 April 2022).
- Daly, M.B.; Pal, T.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hendrix, A.; Hutton, M.L.; et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2. 2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 9 March 2022).
- Berg, J.S.; Agrawal, P.B.; Bailey, D.B., Jr.; Beggs, A.H.; Brenner, S.E.; Brower, A.M.; Cakici, J.A.; Ceyhan-Birsoy, O.; Chan, K.; Chen, F.; et al. Newborn sequencing in genomic medicine and public health. Pediatrics 2017, 139, e20162252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Liu, J.P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef] [PubMed]
- Elliott, L.S.; Henderson, J.C.; Neradilek, M.B.; Moyer, N.A.; Ashcraft, K.C.; Thirumaran, R.K. Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: A prospective pilot randomized controlled trial. PLoS ONE 2017, 12, e0170905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozda, K.; Pacanowski, M.A. Clinical trial designs to support clinical utility of pharmacogenomic testing. Pharmacotherapy 2017, 37, 1000–1004. [Google Scholar] [CrossRef]
- United States Food and Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 14 April 2021).
- Clinical Pharmacogenetics Implementation Consortium. Available online: https://cpicpgx.org (accessed on 17 December 2020).
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef]
- Van Hout, C.V.; Tachmazidou, I.; Backman, J.D.; Hoffman, J.D.; Liu, D.; Pandey, A.K.; Gonzaga-Jauregui, C.; Khalid, S.; Ye, B.; Banerjee, N.; et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 2020, 586, 749–756. [Google Scholar] [CrossRef]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef]
- Buchanan, A.H.; Lester Kirchner, H.; Schwartz, M.L.B.; Kelly, M.A.; Schmidlen, T.; Jones, L.K.; Hallquist, M.L.G.; Rocha, H.; Betts, M.; Schwiter, R.; et al. Clinical outcomes of a genomic screening program for actionable genetic conditions. Genet. Med. 2020, 22, 1874–1882. [Google Scholar] [CrossRef]
- Sugarman, E.A.; Cullors, A.; Centeno, J.; Taylor, D. Contribution of pharmacogenetic testing to modeled medication change recommendations in a long-term care population with polypharmacy. Drugs Aging 2016, 33, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Blasco-Fontecilla, H. Clinical utility of pharmacogenetic testing in children and adolescents with severe mental disorders. J. Neural Transm. 2019, 126, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Vandenbroucke, J.P.; von Elm, E.; Altman, D.G.; Gøtzsche, P.C.; Mulrow, C.D.; Pocock, S.J.; Poole, C.; Schlesselman, J.J.; Egger, M.; Strobe Initiative. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): Explanation and elaboration. PLoS Med. 2007, 4, e297. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Human Genome Variation Society. Sequence Variant Nomenclature. Available online: https://varnomen.hgvs.org (accessed on 11 September 2020).
- Clinical Pharmacogenetics Implementation Consortium. Genes/Drugs. Available online: https://cpicpgx.org/genes-drugs/ (accessed on 14 December 2020).
- United States Census Bureau. QuickFacts: Kentucky. Available online: https://www.census.gov/quickfacts/KY (accessed on 14 April 2021).
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taliun, D.; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention: National Center for Health Statistics. Life Expectancy at Birth by State 2018. Available online: https://www.cdc.gov/nchs/pressroom/sosmap/life_expectancy/life_expectancy.htm (accessed on 6 February 2022).
- Centers for Disease Control and Prevention: Division for Heart Disease and Stroke Prevention. Heart Disease Death Rates, Total Population Ages 35+. Available online: https://www.cdc.gov/dhdsp/maps/national_maps/hd_all.htm (accessed on 6 February 2022).
- Hoskovec, J.M.; Bennett, R.L.; Carey, M.E.; DaVanzo, J.E.; Dougherty, M.; Hahn, S.E.; LeRoy, B.S.; O’Neal, S.; Richardson, J.G.; Wicklund, C.A. Projecting the supply and demand for certified genetic counselors: A workforce study. J. Genet. Couns. 2018, 27, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Raspa, M.; Moultrie, R.; Toth, D.; Haque, S.N. Barriers and facilitators to genetic service delivery models: Scoping review. Interact. J. Med. Res. 2021, 10, e23523. [Google Scholar] [CrossRef]
- Shaw, T.; Metras, J.; Ting, Z.A.L.; Courtney, E.; Li, S.-T.; Ngeow, J. Impact of appointment waiting time on attendance rates at a clinical cancer genetics service. J. Genet. Couns. 2018, 27, 1473–1481. [Google Scholar] [CrossRef]
- Norman, M.L.; Malcolmson, J.; Randall Armel, S.R.; Gillies, B.; Ou, B.; Thain, E.; McCuaig, J.M.; Kim, R.H. Stay at home: Implementation and impact of virtualising cancer genetic services during COVID-19. J. Med. Genet. 2022, 59, 23–27. [Google Scholar] [CrossRef]
- Vivot, A.; Boutron, I.; Ravaud, P.; Porcher, R. Guidance for pharmacogenomic biomarker testing in labels of FDA-approved drugs. Genet. Med. 2015, 17, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.M.; Haidar, C.E.; Wilkinson, M.R.; Crews, K.R.; Baker, D.K.; Kornegay, N.M.; Yang, W.; Pui, C.-H.; Reiss, U.M.; Gaur, A.; et al. PG4KDS: A model for the clinical implementation of pre-emptive pharmacogenetics. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166C, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Papastergiou, J.; Tolios, P.; Li, W.; Li, J. The Innovative Canadian Pharmacogenomic Screening Initiative in Community Pharmacy (ICANPIC) study. J. Am. Pharm. Assoc. 2017, 57, 624–629. [Google Scholar] [CrossRef]
- United States Food and Drug Administration. FDA Drug Safety Communication: New Restrictions, Contraindications, and Dose Limitations for Zocor (Simvastatin) to Reduce the Risk of Muscle Injury. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-new-restrictions-contraindications-and-dose-limitations-zocor (accessed on 9 June 2021).
- Shields, L.B.E.; Fowler, P.; Siemens, D.M.; Lorenz, D.J.; Wilson, K.C.; Hester, S.T.; Honaker, J.T. Standardized warfarin monitoring decreases adverse drug reactions. BMC Fam. Pract. 2019, 20, 151. [Google Scholar] [CrossRef] [Green Version]
- Vonkeman, H.E.; van de Laar, M.A. Nonsteroidal anti-inflammatory drugs: Adverse effects and their prevention. Semin. Arthritis Rheum. 2010, 39, 294–312. [Google Scholar] [CrossRef]
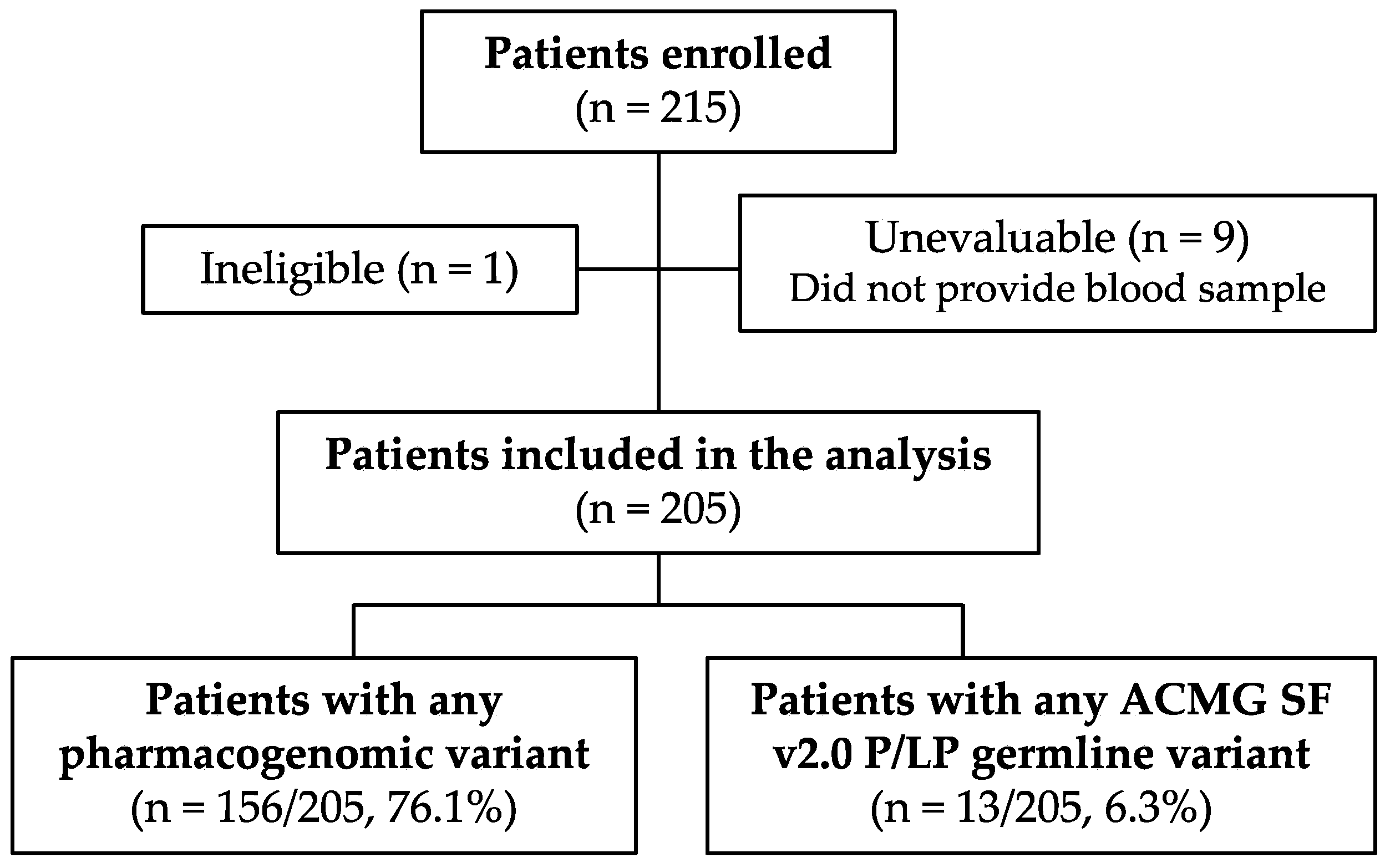
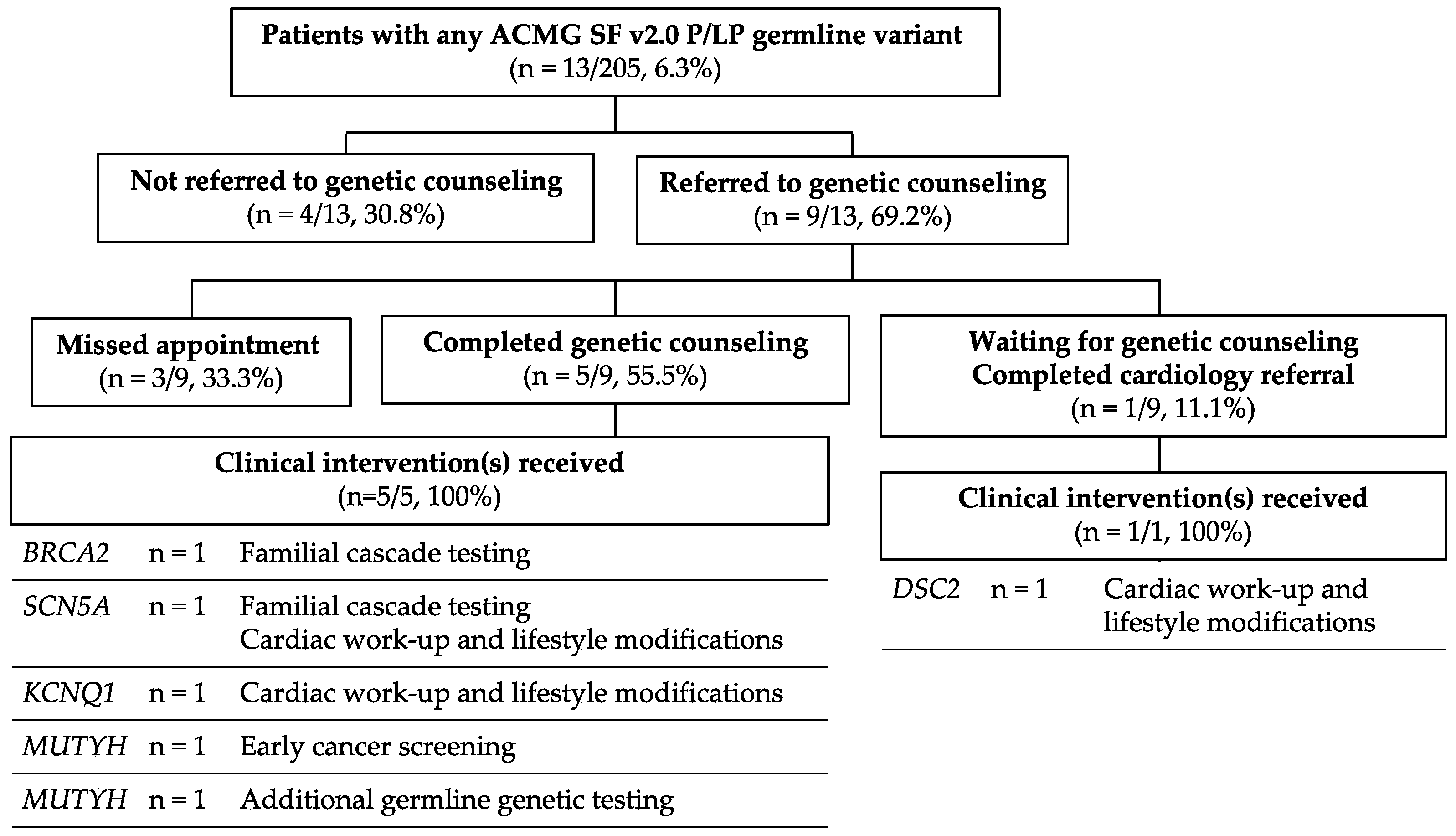
| Characteristic | Patients n (%) |
|---|---|
| Total | 205 |
| Age (median, IQR) | 61 (51–68) |
| Race | |
| AI/AN | 2 (1.0%) |
| Asian | 4 (2.0%) |
| Non-Hispanic Black | 31 (15.1%) |
| Hispanic White | 5 (2.4%) |
| Non-Hispanic White | 163 (79.5%) |
| Gender | |
| Female | 109 (53.2%) |
| Male | 96 (46.8%) |
| Pharmacogenomic Variant Gene | Carrier Frequency | Genotype, n |
|---|---|---|
| Any | 156 (76.1%) | |
| CYP4F2 | 108 (52.7%) | *3/*3: 19 *1/*3: 89 |
| SLCO1B1 | 51 (24.9%) | *15 or *17 a / *15 or *17: 3 *1/*15 or *17: 42 *1/*5: 6 |
| VKORC1 | 29 (14.1%) | *1173C>T/*1173C>T: 3 *1/*1173C>T: 26 |
| CYP2C9 | 19 (9.3%) | *1/*3: 19 |
| CYP3A5 | 10 (4.9%) | *1/*6: 5 *1/*7: 5 |
| DPYD | 10 (4.9%) | *1/*c.2846A>T: 2 *1/*2A: 2 *HapB3/*HapB3: 1 *1/*HapB3: 5 |
| TPMT | 9 (4.4%) | *1/*3A: 6 *1/*3B: 1 *1/*3C: 2 |
| CYP2D6 | 8 (3.9%) | *1/*6: 8 |
| G6PD | 3 (1.5%) | Class III deficiency: 3 |
| Pharmacogenetic Variant | Observed Allele Frequency | Expected Allele Frequency a |
|---|---|---|
| CYP4F2 | ||
| *3 | 0.3098 | 0.4108 |
| SLCO1B1 | ||
| *15 or *17 b | 0.1171 | 0.1214 (*15); 0.0519 (*17) |
| *5 | 0.0146 | 0.0224 |
| VKORC1 | ||
| *-1639G>A | 0.0780 | 0.4643 |
| CYP2C9 | ||
| *3 | 0.0463 | 0.0301 |
| CYP3A5 | ||
| *6 | 0.0122 | 0.0015 |
| *7 | 0.0122 | 0.0000 |
| DPYD | ||
| *c.2846A>T | 0.0048 | 0.0037 |
| *2A | 0.0048 | 0.0079 |
| *HapB3 | 0.0171 | 0.0237 |
| TPMT | ||
| *3A | 0.0146 | 0.0343 |
| *3B | 0.0024 | 0.0027 |
| *3C | 0.0048 | 0.0047 |
| CYP2D6 | ||
| *6 | 0.0195 | 0.0025 |
| G6PD | ||
| A-202A_376G-III | 0.0073 | 0.0–0.034 c |
| Allele Frequency | |||||
|---|---|---|---|---|---|
| Gene and Variant | Observed | Expected a | p-Value | OR (95% CI) | Adjusted p-Value b |
| MUTYH c.1187G>A (p.Gly396Asp) c.536A>G (p.Tyr179Cys) | 0.004878 0.002439 | 0.003027 0.001535 | 0.353 0.468 | 1.61 (0.20, 5.90) 1.59 (0.04, 8.96) | 0.468 0.468 |
| CACNA1S c.4161delC (p.Thr1388Profs*36) c.930delC (p.Trp311Glyfs*23) | 0.002439 0.002439 | Novel c Novel c | - - | - - | - - |
| KCNQ1 c.1075C>T (p.Gln359*) c.1394-1G>T | 0.002439 0.002439 | 1/264,690 d 1/251,392 | 0.003 0.003 | 647.16 (8.22, 4.50 × 1015) 614.65 (7.81, 4.50 × 1015) | 0.019 0.019 |
| APOB c.2477_2478dupTT (p.Leu827Phefs*37) | 0.002439 | Rare e | - | - | - |
| BRCA2 c.2517C>A (p.Tyr839*) | 0.002439 | Rare e | - | - | - |
| DSC2 c.2184dupT (p.Pro729Serfs*2) | 0.002439 | Rare e | - | - | - |
| LDLR c.858C>A (p.Ser286Arg) | 0.002439 | 0.001077 d | 0.358 | 2.27 (0.06, 12.81) | 0.468 |
| SCN5A c.4877G>A (p.Arg1626His) | 0.002439 | 10/251,192 | 0.018 | 61.41 (1.41, 431.59) | 0.071 |
| SDHB c.418G>T (p.Val140Phe) | 0.002439 | 3/251,418 | 0.006 | 204.90 (3.89, 2696.96) | 0.033 |
| Variant Pharmacogene Carriers (n) | Drug Class | Patients Prescribed Drug in Class (n) | Specific Drug Prescribed | Patients Prescribed Specific Drug (n) | Toxicity? (n) | Drug Change? (n) |
|---|---|---|---|---|---|---|
| CYP2C9 (19) | AC | 1 | Warfarin | 0 | - | - |
| NSAID | 7 | Meloxicam Ibuprofen | 1 3 | 0 2 a | 0 0 | |
| AED | 2 | Phenytoin Fosphenytoin | 0 0 | - - | - - | |
| CYP2D6 (8) | Narcotic | 1 | Codeine | 0 | - | - |
| TCA | 1 | Amitriptyline | 1 | 0 | 0 | |
| SSRI | 2 | Paroxetine | 0 | - | - | |
| CYP4F2 (108) | AC | 9 | Warfarin | 1 | 0 | 0 |
| SLCO1B1 (51) | Statin | 27 | Simvastatin | 4 | 0 | 3 b |
| VKORC1 (29) | AC | 1 | Warfarin | 0 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutchcraft, M.L.; Zhang, S.; Lin, N.; Gottschalk, G.L.; Keck, J.W.; Belcher, E.A.; Sears, C.; Wang, C.; Liu, K.; Dietz, L.E.; et al. Real-World Evaluation of a Population Germline Genetic Screening Initiative for Family Medicine Patients. J. Pers. Med. 2022, 12, 1297. https://doi.org/10.3390/jpm12081297
Hutchcraft ML, Zhang S, Lin N, Gottschalk GL, Keck JW, Belcher EA, Sears C, Wang C, Liu K, Dietz LE, et al. Real-World Evaluation of a Population Germline Genetic Screening Initiative for Family Medicine Patients. Journal of Personalized Medicine. 2022; 12(8):1297. https://doi.org/10.3390/jpm12081297
Chicago/Turabian StyleHutchcraft, Megan Leigh, Shulin Zhang, Nan Lin, Ginny Lee Gottschalk, James W. Keck, Elizabeth A. Belcher, Catherine Sears, Chi Wang, Kun Liu, Lauren E. Dietz, and et al. 2022. "Real-World Evaluation of a Population Germline Genetic Screening Initiative for Family Medicine Patients" Journal of Personalized Medicine 12, no. 8: 1297. https://doi.org/10.3390/jpm12081297
APA StyleHutchcraft, M. L., Zhang, S., Lin, N., Gottschalk, G. L., Keck, J. W., Belcher, E. A., Sears, C., Wang, C., Liu, K., Dietz, L. E., Pickarski, J. C., Wei, S., Cardarelli, R., DiPaola, R. S., & Kolesar, J. M. (2022). Real-World Evaluation of a Population Germline Genetic Screening Initiative for Family Medicine Patients. Journal of Personalized Medicine, 12(8), 1297. https://doi.org/10.3390/jpm12081297