
A Systems Biology Approach to Investigate Kinase Signal Transduction Networks That Are Involved in Triple Negative Breast Cancer Resistance to Cisplatin

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Proliferation Assays
2.3. Determination of Baseline Protein Expression
2.4. Reverse-Phase Protein Array
2.5. Bayesian Molecular Response Analysis (BMRA)
2.6. Clustering
2.7. Statistical Analysis
3. Results
3.1. Classification of Cisplatin Response in a Panel of TNBC Cell Lines
3.2. Combination of PI3K/AKT Inhibitors and Cisplatin Are Effective in Double TP53/PIK3CA Mutant TNBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Colaprico, A.; Silva, T.C.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.L.; Balko, J.M.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat. Commun. 2021, 12, 6276. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2021, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Lynce, F.; Nunes, R. Role of Platinums in Triple-Negative Breast Cancer. Curr. Oncol. Rep. 2021, 23, 50. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in our understanding of the molecular mechanisms of action of cisplatin in cancer therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Lee, M.J.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef]
- Martin, A.; Clynes, M. Acid phosphatase: Endpoint for in vitro toxicity tests. Vitro Cell. Dev. Biol. 1991, 27, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Lu, Y.; Gonzalez-Angulo, A.M.; Carey, M.S.; Myhre, S.; Ju, Z.; Davies, M.A.; Liu, W.; Coombes, K.; Meric-Bernstam, F.; et al. A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin. Proteom. 2010, 6, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Halasz, M.; Kholodenko, B.N.; Kolch, W.; Santra, T. Integrating network reconstruction with mechanistic modeling to predict cancer therapies. Sci. Signal. 2016, 9, ra114. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Bhattacharyya, R.; Bauri, A.K.; Patro, B.S.; Chattopadhyay, S. DNA damage dependent activation of checkpoint kinase-1 and mitogen-activated protein kinase-p38 are required in malabaricone C-induced mitochondrial cell death. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, D.O.; Brinkman, E.K.; Marteijn, J.A.; Medema, R.H.; Kanaar, R.; Smits, V.A.J. UV-induced G2 checkpoint depends on p38 MAPK and minimal activation of ATR-Chk1 pathway. J. Cell Sci. 2013, 126, 1923–1930. [Google Scholar] [CrossRef]
- Shin, I.; Yakes, F.M.; Rojo, F.; Shin, N.-Y.; Bakin, A.V.; Baselga, J.; Arteaga, C.L. PKB/Akt mediates cell-cycle progression by phosphorylation of p27Kip1 at threonine 157 and modulation of its cellular localization. Nat. Med. 2002, 8, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Li, C.H.; Karantza, V.; Aktan, G.; Lala, M. Current treatment landscape for patients with locally recurrent inoperable or metastatic triple-negative breast cancer: A systematic literature review. Breast Cancer Res. 2019, 21, 143. [Google Scholar] [CrossRef]
- Georgy, J.T.; Singh, A.; John, A.O.; Joel, A.; Andrews, A.G.; Thumaty, D.B.; Rebekah, G.; Sigamani, E.; Chandramohan, J.; Manipadam, M.T.; et al. Pathological response and clinical outcomes in operable triple-negative breast cancer with cisplatin added to standard neoadjuvant chemotherapy. Klin. Onkol. 2021, 34, 49–55. [Google Scholar]
- Egger, S.J.; Chan, M.M.K.; Luo, Q.; Wilcken, N. Platinum-containing regimens for triple-negative metastatic breast cancer. Cochrane Database Syst. Rev. 2020, 10, CD013750. [Google Scholar] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 10, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Hill, D.P.; Harper, A.; Malcolm, J.; McAndrews, M.S.; Mockus, S.M.; Patterson, S.E.; Reynolds, T.; Baker, E.J.; Bult, C.J.; Chesler, E.J.; et al. Cisplatin-resistant triple-negative breast cancer subtypes: Multiple mechanisms of resistance. BMC Cancer 2019, 19, 1039. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef]
- Machl, A.; Wilker, E.W.; Tian, H.; Liu, X.; Schroeder, P.; Clark, A.; Huck, B.R. M2698 is a potent dual-inhibitor of p70S6K and Akt that affects tumor growth in mouse models of cancer and crosses the blood-brain barrier. Am. J. Cancer Res. 2016, 6, 806–818. [Google Scholar]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef]
- Yuan, Y.; Wen, W.; Yost, S.E.; Xing, Q.; Yan, J.; Han, E.S.; Mortimer, J.; Yim, J.H. Combination therapy with BYL719 and LEE011 is synergistic and causes a greater suppression of p-S6 in triple negative breast cancer. Sci. Rep. 2019, 9, 7509. [Google Scholar] [CrossRef]
- Hatem, R.; El Botty, R.; Chateau-Joubert, S.; Servely, J.L.; Labiod, D.; de Plater, L.; Assayag, F.; Coussy, F.; Callens, C.; Vacher, S.; et al. Targeting mTOR pathway inhibits tumor growth in different molecular subtypes of triple-negative breast cancers. Oncotarget 2016, 7, 48206–48219. [Google Scholar] [CrossRef]
- Gohr, K.; Hamacher, A.; Engelke, L.H.; Kassack, M.U. Inhibition of PI3K/Akt/mTOR overcomes cisplatin resistance in the triple negative breast cancer cell line HCC38. BMC Cancer 2017, 17, 711. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Reddy, P.G.; Goberdhan, A.; Walsh, C.; Dao, S.; Ngai, I.; Chou, T.C.; O-charoenrat, P.; Levine, A.J.; Rao, P.H.; et al. p53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes Dev. 2002, 16, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Astanehe, A.; Arenillas, D.; Wasserman, W.W.; Leung, P.C.K.; Dunn, S.E.; Davies, B.R.; Mills, G.B.; Auersperg, N. Mechanisms underlying p53 regulation of PIK3CA transcription in ovarian surface epithelium and in ovarian cancer. J. Cell Sci. 2008, 121, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.; Ray, P. p53 Loses grip on PIK3CA expression leading to enhanced cell survival during platinum resistance. Mol. Oncol. 2016, 10, 1283–1295. [Google Scholar] [CrossRef]
- Chen, X.; Guo, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xu, Y.; Xie, Y. Co-mutation of TP53 and PIK3CA in residual disease after neoadjuvant chemotherapy is associated with poor survival in breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.Y.; Bian, L.; Nicklawsky, A.G.; Krinsky, A.L.; Brunetti, T.; Woolaver, R.A.; Wang, X.; Chen, Z.; Young, C.D.; Gao, D.; et al. Deletion of p53 and hyper-activation of pik3ca in keratin-15+ stem cells lead to the development of spontaneous squamous cell carcinoma. Int. J. Mol. Sci. 2020, 21, 6585. [Google Scholar] [CrossRef] [PubMed]
- Croessmann, S.; Wong, H.Y.; Zabransky, D.J.; Chu, D.; Rosen, D.M.; Cidado, J.; Cochran, R.L.; Dalton, W.B.; Erlanger, B.; Cravero, K.; et al. PIK3CA mutations and TP53 alterations cooperate to increase cancerous phenotypes and tumor heterogeneity. Breast Cancer Res. Treat. 2017, 162, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohée, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, J.; Zhong, Y.; Geng, R.; Ji, Y.; Guan, Q.; Hong, C.; Wei, Y.; Min, N.; Qi, A.; et al. PIK3CA mutation confers resistance to chemotherapy in triple-negative breast cancer by inhibiting apoptosis and activating the PI3K/AKT/mTOR signaling pathway. Ann. Transl. Med. 2021, 9, 410. [Google Scholar] [CrossRef] [PubMed]
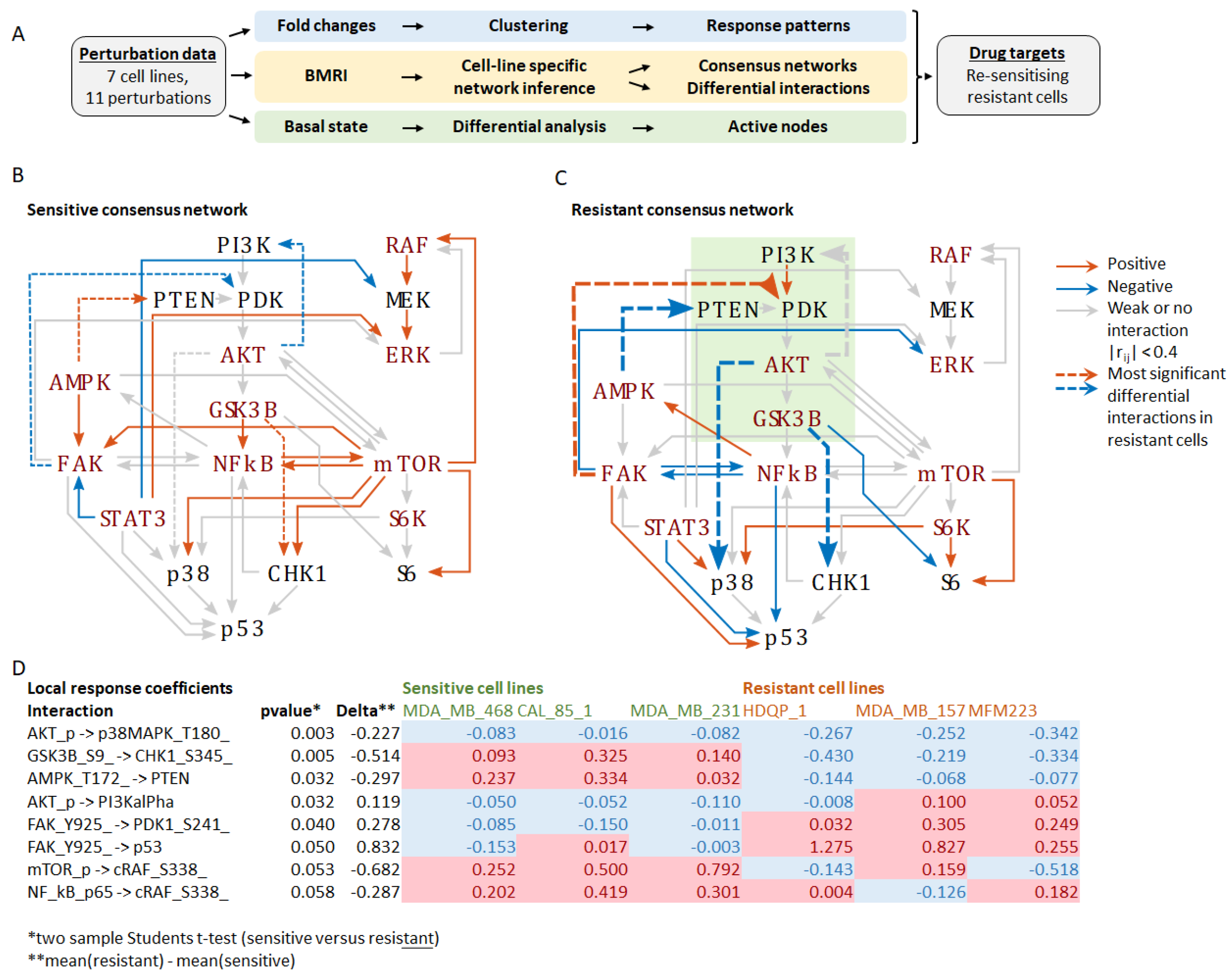
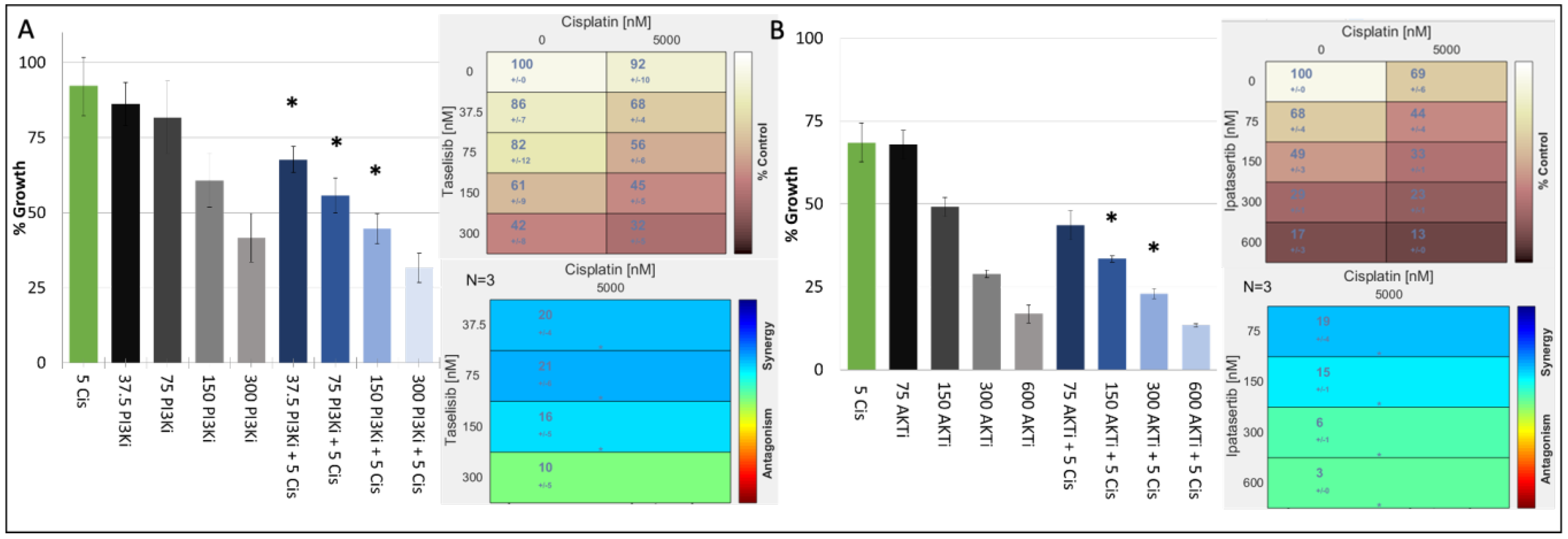

{kind=link}
{kind=link}
| Target | Inhibitor | Concentration (nM) | Duration of Treatment |
|---|---|---|---|
| AKT | Ipatasertib | 500 | 1 h |
| AMPK | AICAR (Acadesine) * | 1 | 1 h |
| AMPK | Dorsomorphin ** | 20,000 | 1 h |
| RAF | TAK-632 | 20,000 | 1 h |
| FAK | PF-00562271 | 100 | 1 h |
| GSK3B | CHIR-98014 | 10,000 | 1 h |
| MEK | U0126 | 10,000 | 1 h |
| mTOR | Everolimus | 1 | 1 h |
| NFkB | QNZ (EVP4593) | 1000, 5000 | 1 h |
| p70S6K | PF-4708671 | 10,000 | 1 h |
| STAT3 | Stattic | 5000 | 1 h |
| DNA | Cisplatin | 5000 | 1 h |
| Cell Lines | Triple Negative Subtype | Cisplatin IC50 (µM) | Cisplatin (% Inhibition @ 10 µM) | Cisplatin Response |
|---|---|---|---|---|
| HDQ-P1 | Basal-like 2 | 6.33 ± 0.46 | 53.4 ± 2.3 | Resistant |
| MDA-MB-157 | Mesenchymal stem-like | 8.90 ± 0.83 | 47.8 ± 16.1 | Resistant |
| MFM223 | LAR | >10 | 23.3 ± 2.1 | Resistant |
| CAL120 | Mesenchymal-like | 12.3 ± 3.6 | 44.6 ± 2.3 | Resistant |
| MCF10A | Normal | 5.25 ± 0.55 | 69.5 ± 2.6 | - |
| MDA-MB-468 | Basal-like 1 | 0.23 ± 0.03 | 99.5 ± 0.5 | Sensitive |
| HCC1143 | Basal-like 1 | 1.07 ± 0.32 | 95.9 ± 2.1 | Sensitive |
| MDA-MB-231 | Mesenchymal stem-like | 2.13 ± 0.42 | 77.5 ± 8.4 | Sensitive |
| CAL-85-1 | Basal-like 2 | 0.47 ± 0.04 | 96.6 ± 1.0 | Sensitive |
| Cell Line | PIK3CA Mutation | TP53 Mutation | Cisplatin IC50 μM | M2698 IC50 μM | CHIR-98014 % Inhibition @ 1 μM | Taselisib IC50 μM | Ipataseritib IC50 μM |
|---|---|---|---|---|---|---|---|
| Cisplatin | P70S6 Kinase Inhibitor | GSK3B Inhibitor | PI3K Inhibitor (αδ Specific) | AKT Inhibitor | |||
| BT20 | H1047R/P539R | K132Q | 0.43 ± 0.08 | n/a | n/a | 0.11 ± 0.03 | n/a |
| CAL-51 | E542K | WT | 2.10 ± 0.60 | n/a | n/a | 0.03 ± 0.02 | n/a |
| CAL85-1 | WT | K132E | 0.47 ± 0.04 | 0.94 ± 0.65 | n/a | n/a | n/a |
| HCC1143 | WT | R248Q | 1.07 ± 0.32 | 1.90 ± 0.58 | −5 ± 6% | n/a | n/a |
| HDQ-P1 | WT | R213 * | 6.33 ± 0.46 | 7.34 ± 3.40 | 37 ± 10% | 0.67 ± 0.16 | n/a |
| MDA-MB-157 | WT | FS Del | 8.90 ± 0.83 | 4.11 ± 1.11 | 20 ± 21% | >5 uM | n/a |
| MDA-MB-231 | WT | R280K | 2.13 ± 0.42 | 2.00 ± 0.45 | 49 ± 4% | n/a | n/a |
| MDA-MB-468 | WT | R273H | 0.23 ± 0.03 | 0.38 ± 0.91 | 82 ± 7% | n/a | n/a |
| MFM-223 | H1047R | K132R | >10 | 0.11 ± 0.02 | 17% ± 6 | 0.17 ± 0.06 | 0.27 ± 0.15 |
| Cisplatin ± Taselisib-PI3K Inhibitor | ||||||
|---|---|---|---|---|---|---|
| Cell Line | Cisplatin IC50 (μM) | PIK3CA Mutation | TP53 Mutation | Best Loewe Synergy | PI3K Inhi#bitor conc. (nM) | Cisplatin (nM) |
| MFM223 | >10 | H1047R | K132R | 21 (±6) | 75 | 5000 |
| HDQP1 | 6.4 | WT | R213 * | 9 (±3) | 25 | 1250 |
| MDAMB157 | 8 | WT | FS Del | 13 (±5) | 100 | 1250 |
| CAL51 | 2.1 | E542K | WT | 12 (±2) | 5 | 600 |
| BT20 | 0.43 | H1047R/ P539R | K132Q | 11 (±2) | 150 | 200 |
| Cisplatin ± Ipatasertib-AKT Inhibitor | ||||||
| Cell Line | Cisplatin IC50 (μM) | PIK3CA Mutation | TP53 mutation | Best Loewe synergy | AKTi conc. (nM) | Cisplatin (nM) |
| MFM223 | >10 | H1047R | K132R | 19 (±4) | 75 | 5000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, N.; Browne, A.; Ivers, L.; Santra, T.; Cremona, M.; Hennessy, B.T.; O’Donovan, N.; Crown, J.; Kolch, W.; Fey, D.; et al. A Systems Biology Approach to Investigate Kinase Signal Transduction Networks That Are Involved in Triple Negative Breast Cancer Resistance to Cisplatin. J. Pers. Med. 2022, 12, 1277. https://doi.org/10.3390/jpm12081277
Mukherjee N, Browne A, Ivers L, Santra T, Cremona M, Hennessy BT, O’Donovan N, Crown J, Kolch W, Fey D, et al. A Systems Biology Approach to Investigate Kinase Signal Transduction Networks That Are Involved in Triple Negative Breast Cancer Resistance to Cisplatin. Journal of Personalized Medicine. 2022; 12(8):1277. https://doi.org/10.3390/jpm12081277
Chicago/Turabian StyleMukherjee, Nupur, Alacoque Browne, Laura Ivers, Tapesh Santra, Mattia Cremona, Bryan T. Hennessy, Norma O’Donovan, John Crown, Walter Kolch, Dirk Fey, and et al. 2022. "A Systems Biology Approach to Investigate Kinase Signal Transduction Networks That Are Involved in Triple Negative Breast Cancer Resistance to Cisplatin" Journal of Personalized Medicine 12, no. 8: 1277. https://doi.org/10.3390/jpm12081277
APA StyleMukherjee, N., Browne, A., Ivers, L., Santra, T., Cremona, M., Hennessy, B. T., O’Donovan, N., Crown, J., Kolch, W., Fey, D., & Eustace, A. J. (2022). A Systems Biology Approach to Investigate Kinase Signal Transduction Networks That Are Involved in Triple Negative Breast Cancer Resistance to Cisplatin. Journal of Personalized Medicine, 12(8), 1277. https://doi.org/10.3390/jpm12081277