
Global Globin Network Consensus Paper: Classification and Stratified Roadmaps for Improved Thalassaemia Care and Prevention in 32 Countries

 ,
,  , , , , , ,
, , , , , ,  , ,
, ,  ,
, 
Abstract
1. Introduction
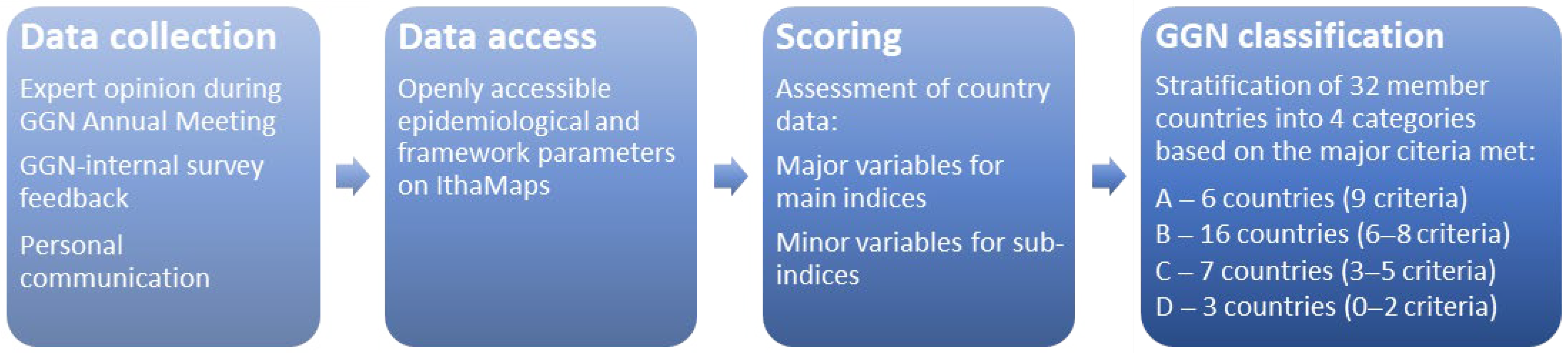
2. Materials and Methods
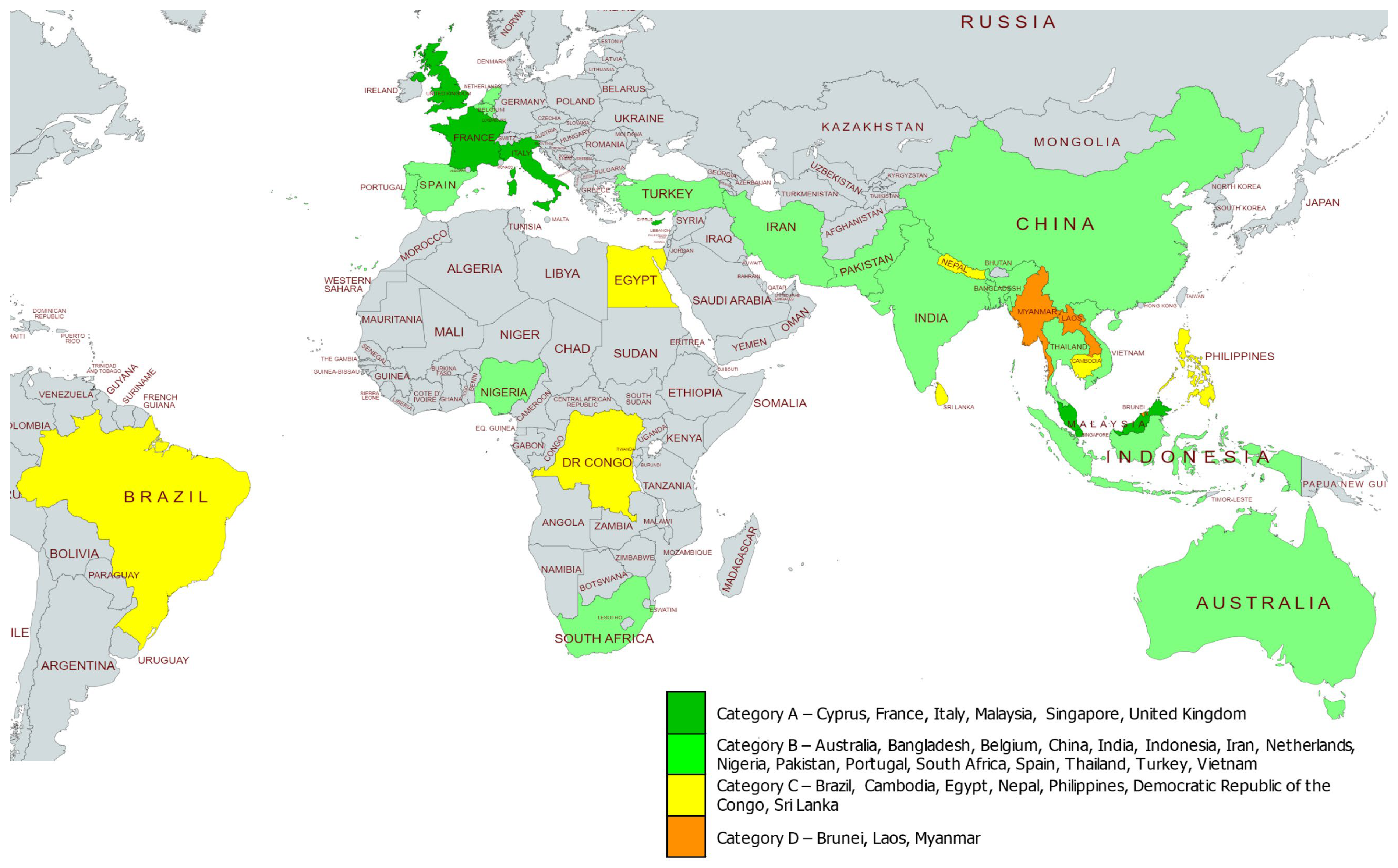
3. Results
3.1. Category A

{kind=link}
{kind=link}
| Country | Prevalence of Carriers | Ethnic Group(s) Affected | Policy Applied in the Country | ||||
|---|---|---|---|---|---|---|---|
| Prevention Programme | Prenatal/Antenatal Screening | Sickle Cell Disease Newborn Screening | Thalassaemia Registry | ||||
| National Level | Local/District Level | ||||||
| Cyprus | βThal 12% (Greek Cypriots) αThal 20% (Greek Cypriots) [23,38,39] | Greek Cypriot Turkish Cypriot [23,39] | Yes [23,39] | Yes [23,39] | Yes [23,39] | No | National Thalassaemia Registry [39] |
| France | βThal (0.7%) SCD (0.7%) HbE (0.15%) HbC (0.2%) [40,41] | Guadeloupe | No [21] | Yes [21] | |||
| Italy | βThal (6%) αThal (6%) SCD (2%) HbC (1%) HbE (0.2%) | Italian, Sardinian [27] | Yes [21] | Yes [21] | Yes [21,27] | Yes [21] | National Thalassaemia Registry [21,27] |
| Malaysia | βThal (4.5%), αThal (4.9%), HbE (5.5%) [42,43] | Malay (62.0%) Kadazan-Dusun (14.0%) Chinese (13.0%) Indian (1.0%) Others (10.0%) [33,34] | Yes [33] | Yes | Yes [42] | No | Yes [Malaysian Thalassaemia Registry] [33,34] |
| Singapore | βThal (1.6%), αThal (incl. HbCS) (5.5%) HbE (1.7%) [44] | αThal/βThal Chinese (6.4%/2.7%) Malay (4.8%/6.3%) Indian (5.2%/0.7%) [44] | Yes [44] | National Thalassaemia Registry [29] | |||
| United Kingdom | βThal (0.44%) SCD (2.5%) αThal (2.5%) HbC (0.13%) [40] | Irish, Anglo-Saxon and multi-ethnic | Yes [35] | Yes [21,36] | Yes [35] | Yes [37] | |
3.2. Category B
| Country | Prevalence of Carriers | Ethnic Group(s) Most Affected | Policy Applied in the Country | ||||
|---|---|---|---|---|---|---|---|
| Prevention Programme | Prenatal/Antenatal Screening | Sickle Cell Disease Screening | Thalassaemia Registry | ||||
| National Level | Local/District Level | ||||||
| Australia | βThal (0.4%) HbE (0.4%) [22,48] | Yes [22] | Prenatal (Yes), Antenatal (No) [22] | Yes | |||
| Bangladesh | α/βThal (4.1–12%) HbE (6.1%) [49,50] | Bengali, Marma, Khyang | No | No | No | ||
| Belgium | SCD (0.42%) βThal (0.28%) HbE (0.02%) HbC (0.02%) [40,41,51] | Northern European (lowest risk) | No [35] | No | No [51] | Yes | |
| China, Guanxi | αThal (3.54%) βThal (6.78%) HbE (0.42%) [22] | Yes (Regional) [35] | Yes [52] | No [35] | Yes [53] | ||
| India | αThal (41.0%) βThal (3.9%) HbE (1.0%) [22] 1:8 of all Thal carriers worldwide; regional range 0.6–15% [47] | Gujarat (10–15%), Tamil Nadu (8.5%), Punjab (6.5%) [47] | Yes [52] | Yes [52,54] | Yes [35] | Yes [52] | |
| Indonesia | αThal (10.9%) βThal (5%) HbE (6%) [22] | Malay, Javanese, Aceh, Batak, Sundanese, Padang, Betawi, South Celebes | Yes [22] | Yes [22] | Yes (National) | ||
| Iran | βThal (6%) SCD (1%) αThal (30%) [40,55] | Yes (National) [56] | Yes [56] | No [35] | Yes [57] | ||
| Netherlands | SCD (0.18%) βThal (0.4%) HbE (0.07%) HbC (0.1%) αThal (3.6%) [40,41] | Dutch | Yes [35] | Prenatal (No) [58] Antenatal (Yes) [21] | Yes | Yes [21] | |
| Nigeria | SCD (25.0%) [59] | Yoruba | Yes | Yes | Yes | No | |
| Pakistan | βThal (5%) αThal (2.4%) SCD (0.27%) | Yes | Yes | ||||
| Portugal | βThal (1.63%) SCD (0.12%) HbE (0.002%) HbC (0.01%) [41,60] | Portuguese | No [35] | No | No [35] | ||
| South Africa | βThal (2–20%) Indian/Mediterranean SCD (≤20%) Central and West African αThal (≤30%) single alpha deletion | Mediterranean, Indian, Central and West African | No | No | Yes | No | No |
| Spain | βThal (1.64%) SCD (0.3%) HbE (0.002%) HbC (0.03%) [40,41] | Spanish | No [35] | No [35] | Yes [37] | ||
| Thailand | βThal (3–9%) HbE (10–53%) HbCS (1–8%) αThal (20–30%) HbE (33% [22] | Thais [22] | Yes | - | Yes [22,52] | No | Yes |
| Turkey | βThal (2.2%) SCD (0.44%) αThal (2%) HbE (0.002%) HbC (0.001%) [40] | Turkish | Yes [61] | - | Yes [61] | No [35] | Yes [62] |
| Vietnam | αThal (11.7%) βThal (2.6%) HbE (1%) [22] | Kinh Muong Tay | Yes | No [63] | |||
3.3. Category C
| Country | Prevalence of Carriers | Ethnic Group(s) Most Affected | Policy Applied in the Country | ||||
|---|---|---|---|---|---|---|---|
| Prevention Programme | Prenatal/Antenatal Screening | Sickle Cell Disease Screening | Thalassaemia Registry | ||||
| National Level | Local/District Level | ||||||
| Brazil | βThal (6%) SCD (1%) αThal (30%) [40,70] | Brazilian | No | Yes [71] | Yes | ||
| Cambodia | βThal (0.18%) αThal (18.27%) HbE (19.93%) [72] | Khmer with regional differences | No | No | No | ||
| Egypt | βThal (5.3%) SCD (2.54%) αThal (9.25%) [40,73] | Yes | No | Yes [52] | No [35] | ||
| Nepal | HbE (4.0%) [74] | High case counts in Bheri Zonal Hospital, Nepalgunj; low in Bharatpur Hospital, Chitwan | No | ||||
| Philippines | αThal (20.4%) βThal (1.2%) HbE (0.4%) [22] | No | No | No | Yes | No | |
| Democratic Republic of the Congo | SCD (24%, inferred from 2% homozygote births) αThal (1% non-deletional, inferred from 49% carriers among SCD patients) [75,76] | No | |||||
| Sri Lanka | αThal (6.5%) βThal (2.5%) HbE (2.5%) [22] | Yes [52] | Yes | Yes [77] | |||
3.4. Category D
| Country | Prevalence of Carriers | Ethnic Group(S) Most Affected | Policy Applied in the Country | ||||
|---|---|---|---|---|---|---|---|
| Prevention Programme | Prenatal/Antenatal Screening | Sickle Cell Disease Screening | Thalassaemia Registry | ||||
| National Level | Local/District Level | ||||||
| Brunei | αThal (4.3%) βThal (2%) HbE (0%) [22] | Malay | No | No | No | Yes | |
| Laos | αThal (26.8%) [80] For pregnant Laotians: HbE (30.1%) α0Thal (8.6%) | Yes | Yes | Yes | |||
| Myanmar | αThal (10–56.9%) βThal (0.5–4.1%) [78] | No | |||||
4. Discussion
4.1. Screening and Disease-Prevention Strategies
4.2. Education and Public Awareness
4.3. Reliable Data for Reliable Disease Control
5. Recommendations and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Modell, B.; Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Stephanou, C.; Tamana, S.; Minaidou, A.; Papasavva, P.; Kleanthous, M.; Kountouris, P. Genetic Modifiers at the Crossroads of Personalised Medicine for Haemoglobinopathies. J. Clin. Med. 2019, 8, 1927. [Google Scholar] [CrossRef] [PubMed]
- Rosnah, B.; Rosline, H.; Zaidah, A.W.; Noor Haslina, M.N.; Marini, R.; Shafini, M.Y.; Nurul Ain, F.A. Detection of common deletional alpha-thalassemia spectrum by molecular technique in Kelantan, northeastern Malaysia. ISRN Hematol. 2012, 2012, 462969. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An interactive database for haemoglobin variations and epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef]
- Weatherall, D.J. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010, 115, 4331–4336. [Google Scholar] [CrossRef]
- Williams, T.N.; Weatherall, D.J. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb. Perspect. Med. 2012, 2, a011692. [Google Scholar] [CrossRef]
- Burn, J.; Watson, M. The Human Variome Project. Hum. Mutat. 2016, 37, 505–507. [Google Scholar] [CrossRef][Green Version]
- Massie, R.J.; Delatycki, M.B. Reducing the burden of inherited disease: The Human Variome Project. Med. J. Aust. 2010, 193, 430–431. [Google Scholar] [CrossRef]
- Smith, T.D.; Vihinen, M.; Human Variome, P. Standard development at the Human Variome Project. Database 2015, 2015, bav024. [Google Scholar] [CrossRef]
- Oetting, W.S.; Robinson, P.N.; Greenblatt, M.S.; Cotton, R.G.; Beck, T.; Carey, J.C.; Doelken, S.C.; Girdea, M.; Groza, T.; Hamilton, C.M.; et al. Getting ready for the Human Phenome Project: The 2012 forum of the Human Variome Project. Hum. Mutat. 2013, 34, 661–666. [Google Scholar] [CrossRef]
- Yusof, W.; Halim-Fikri, H.; Zilfalil, B.A. The Human Variome Project and Global Globin Challenge. Pak. Pediatr. J. 2017, 41, 202–206. [Google Scholar]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Robinson, H.M. Increasing the involvement of diverse populations in genomics-based health care-lessons from haemoglobinopathies. J. Community Genet. 2017, 8, 311–318. [Google Scholar] [CrossRef][Green Version]
- Stephanou, C.; Kountouris, P.; Lederer, C.W.; Kleanthous, M. Variant Curation: Overview and Challenges. Hemoglobin 2019, 43, 325. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Tamana, S.; Minaidou, A.; Xenophontos, M.; Angastiniotis, M.; Eleftheriou, A.; Zilfalil, B.A.; Ramesar, R.S.; et al. The ITHANET-Human Variome Project: Moving Functional Annotation Forward. Hemoglobin 2019, 43, 327. [Google Scholar] [CrossRef]
- Preston, C.G.; Wright, M.W.; Madhavrao, R.; Harrison, S.M.; Goldstein, J.L.; Luo, X.; Wand, H.; Wulf, B.; Cheung, G.; Mandell, M.E.; et al. ClinGen Variant Curation Interface: A variant classification platform for the application of evidence criteria from ACMG/AMP guidelines. Genome Med. 2022, 14, 6. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Traeger-Synodinos, J.; Bento, C.; Harteveld, C.L.; Fylaktou, E.; Koopmann, T.T.; Halim-Fikri, H.; Michailidou, K.; et al. Adapting the ACMG/AMP variant classification framework: A perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Hum. Mutat. 2021, 1–8. [Google Scholar] [CrossRef]
- Zilfalil, B.A. Global Globin 2020 Challenge: The Past, Present and Future. Hemoglobin 2019, 43, 302. [Google Scholar] [CrossRef]
- Tekola-Ayele, F.; Rotimi, C.N. Translational Genomics in Low- and Middle-Income Countries: Opportunities and Challenges. Public Health Genom. 2015, 18, 242–247. [Google Scholar] [CrossRef]
- Aguilar Martinez, P.; Angastiniotis, M.; Eleftheriou, A.; Gulbis, B.; Manu Pereira Mdel, M.; Petrova-Benedict, R.; Corrons, J.L. Haemoglobinopathies in Europe: Health & migration policy perspectives. Orphanet J. Rare Dis. 2014, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Weatherall, D.J. Progress Toward the Control and Management of the Thalassemias. Hematol. Oncol. Clin. North Am. 2016, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Kousiappa, I.; Papasavva, T.; Christopoulos, G.; Pavlou, E.; Petrou, M.; Feleki, X.; Karitzie, E.; Phylactides, M.; Fanis, P.; et al. The molecular spectrum and distribution of haemoglobinopathies in Cyprus: A 20-year retrospective study. Sci. Rep. 2016, 6, 26371. [Google Scholar] [CrossRef] [PubMed]
- Skordis, N.; Christou, S.; Koliou, M.; Pavlides, N.; Angastiniotis, M. Fertility in female patients with thalassemia. J. Pediatr. Endocrinol. Metab. 1998, 11 (Suppl. 3), 935–943. [Google Scholar]
- Origa, R.; Galanello, R.; Perseu, L.; Tavazzi, D.; Domenica Cappellini, M.; Terenzani, L.; Forni, G.L.; Quarta, G.; Boetti, T.; Piga, A. Cholelithiasis in thalassemia major. Eur. J. Haematol. 2009, 82, 22–25. [Google Scholar] [CrossRef]
- Kountouris, P.; Michailidou, K.; Christou, S.; Hadjigavriel, M.; Sitarou, M.; Kolnagou, A.; Kleanthous, M.; Telfer, P. Effect of HBB genotype on survival in a cohort of transfusion-dependent thalassemia patients in Cyprus. Haematologica 2021, 106, 2458–2468. [Google Scholar] [CrossRef]
- Cao, A.; Rosatelli, M.C.; Monni, G.; Galanello, R. Screening for thalassemia: A model of success. Obstet. Gynecol. Clin. North Am. 2002, 29, 305–328. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Archer, N.; Bonifazi, F.; Giannuzzi, V.; Kuo, K.H.M.; Maggio, A.; Makani, J.; Manu-Pereira, M.D.M.; Michailidou, K.; et al. The International Hemoglobinopathy Research Network (INHERENT): An international initiative to study the role of genetic modifiers in hemoglobinopathies. Am. J. Hematol. 2021, 96, E416–E420. [Google Scholar] [CrossRef]
- Law, H.Y.; Chee, M.K.; Tan, G.P.; Ng, I.S. The simultaneous presence of α- and β-thalassaemia alleles: A pitfall of thalassaemia screening. Community Genet. 2003, 6, 14–21. [Google Scholar] [CrossRef]
- Ng, I.S.; Ong, J.B.; Tan, C.L.; Law, H.Y. Beta-thalassemia mutations in Singapore--a strategy for prenatal diagnosis. Hum. Genet. 1994, 94, 385–388. [Google Scholar] [CrossRef]
- Ng, I.S.; Ong, J.B.; Tan, C.L.; Law, H.Y. Early prenatal diagnosis of beta-thalassaemia in Singapore. Ann. Acad. Med. Singap. 1996, 25, 779–782. [Google Scholar]
- Kham, S.K.; Quah, T.C.; Loong, A.M.; Tan, P.L.; Fraser, A.; Chong, S.S.; Yeoh, A.E. A molecular epidemiologic study of thalassemia using newborns’ cord blood in a multiracial Asian population in Singapore: Results and recommendations for a population screening program. J. Pediatr. Hematol. Oncol. 2004, 26, 817–819. [Google Scholar]
- Mohd Ibrahim, H.; Muda, Z.; Othman, I.S.; Mohamed Unni, M.N.; Teh, K.H.; Thevarajah, A.; Gunasagaran, K.; Ong, G.B.; Yeoh, S.L.; Muhammad Rivai, A.; et al. Observational study on the current status of thalassaemia in Malaysia: A report from the Malaysian Thalassaemia Registry. BMJ Open 2020, 10, e037974. [Google Scholar] [CrossRef]
- Mohd Ibrahim, H. Malaysian Thalassaemia Registry Report 2018, 1st ed.; Ministry of Health, Ed.; Medical Development Division, Ministry of Health: Kementerian Kesihatan, Malaysia, 2019. [Google Scholar]
- Giordano, P.C.; Harteveld, C.L.; Bakker, E. Genetic epidemiology and preventive healthcare in multiethnic societies: The hemoglobinopathies. Int. J. Environ. Res. Public Health 2014, 11, 6136–6146. [Google Scholar] [CrossRef]
- Dormandy, E.; Reid, E.; Tsianakas, V.; O’Neil, B.; Gill, E.; Marteau, T.M. Offering antenatal sickle cell and thalassaemia screening in primary care: A pre-post evaluation of a brief type of communication skills training. Patient Educ. Couns. 2012, 89, 129–133. [Google Scholar] [CrossRef]
- Noori, T.; Ghazisaeedi, M.; Aliabad, G.M.; Mehdipour, Y.; Mehraeen, E.; Conte, R.; Safdari, R. International Comparison of Thalassemia Registries: Challenges and Opportunities. Acta Inform. Med. 2019, 27, 58–63. [Google Scholar] [CrossRef]
- Kyriacou, K.; Kyrri, A.; Kalogirou, E.; Vasiliades, P.; Angastiniotis, M.; Ioannou, P.A.; Kleanthous, M. Hb Bart’s levels in cord blood and alpha-thalassemia mutations in Cyprus. Hemoglobin 2000, 24, 171–180. [Google Scholar] [CrossRef]
- Bozkurt, G. Results from the north cyprus thalassemia prevention program. Hemoglobin 2007, 31, 257–264. [Google Scholar] [CrossRef]
- Angastiniotis, M.; Eleftheriou, A.; Galanello, R.; Harteveld, C.L.; Petrou, M.; Traeger-Synodinos, J.; Giordano, P.; Jauniaux, E.; Modell, B.; Serour, G. Principles. In Prevention of Thalassaemias and Other Haemoglobin Disorders; Old, J., Ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2013; Volume 1. [Google Scholar]
- Modell, B.; Darlison, M.; Birgens, H.; Cario, H.; Faustino, P.; Giordano, P.C.; Gulbis, B.; Hopmeier, P.; Lena-Russo, D.; Romao, L.; et al. Epidemiology of haemoglobin disorders in Europe: An overview. Scand. J. Clin. Lab. Investig. 2007, 67, 39–69. [Google Scholar] [CrossRef]
- Ngim, C.F.; Lai, N.M.; Ibrahim, H. Counseling for prenatal diagnosis and termination of pregnancy due to thalassemia major: A survey of health care workers’ practices in Malaysia. Prenat. Diagn. 2013, 33, 1226–1232. [Google Scholar] [CrossRef]
- George, E.; Ann, T.J. Genotype-phenotype diversity of beta-thalassemia in Malaysia: Treatment options and emerging therapies. Med. J. Malays. 2010, 65, 256–260. [Google Scholar]
- Lee, S.Y.; Yap, E.S.; Lee, E.Y.; Goh, J.H.; Liu, T.C.; Yip, C. Evaluation of Thalassaemia Screening Tests in the Antenatal and Non-Antenatal Populations in Singapore. Ann. Acad. Med. Singap. 2019, 48, 5–15. [Google Scholar]
- Kountouris, P.; Stephanou, C.; Bento, C.; Fanis, P.; Elion, J.; Ramesar, R.S.; Zilfalil, B.A.; Robinson, H.M.; Traeger-Synodinos, J.; Challenge, H.V.P.G.G.; et al. ITHANET: Information and database community portal for haemoglobinopathies. bioRxiv 2017, 209361. [Google Scholar] [CrossRef]
- Suwannakhon, N.; Pongsawatkul, K.; Seeratanachot, T.; Mahingsa, K.; Pingyod, A.; Bumrungpakdee, W.; Sanguansermsri, T. The shortcut strategy for beta thalassemia prevention. Hematol. Rep. 2018, 10, 7530. [Google Scholar] [CrossRef]
- Thiyagarajan, A.; Bhattacharya, S.; Sharma, N.; Srivastava, A.; Dhar, D.K. Need for a universal thalassemia screening programme in India? A public health perspective. J. Fam. Med. Prim. Care 2019, 8, 1528–1532. [Google Scholar] [CrossRef]
- Hendy, J. Preventation of thalassemia in Australia. Southeast Asian J. Trop. Med. Public Health 1999, 30 (Suppl. 2), 94–96. [Google Scholar]
- Bastola, G.; Acharya, R.; Dhakal, N.; Gupta, U.P. Study of Thalassemia and Haemoglobinopathies in Pokhara, Nepal. J. Clin. Diagn. Res. 2017, 11, 15–18. [Google Scholar] [CrossRef]
- Hossain, M.S.; Raheem, E.; Sultana, T.A.; Ferdous, S.; Nahar, N.; Islam, S.; Arifuzzaman, M.; Razzaque, M.A.; Alam, R.; Aziz, S. Thalassemias in South Asia: Clinical lessons learnt from Bangladesh. Orphanet J. Rare Dis. 2017, 12, 93. [Google Scholar] [CrossRef]
- Gulbis, B.; Ferster, A.; Cotton, F.; Lebouchard, M.P.; Cochaux, P.; Vertongen, F. Neonatal haemoglobinopathy screening: Review of a 10-year programme in Brussels. J. Med. Screen. 2006, 13, 76–78. [Google Scholar] [CrossRef]
- Goonasekera, H.W.; Paththinige, C.S.; Dissanayake, V.H.W. Population Screening for Hemoglobinopathies. Annu. Rev. Genom. Hum. Genet. 2018, 19, 355–380. [Google Scholar] [CrossRef] [PubMed]
- Dharssi, S.; Wong-Rieger, D.; Harold, M.; Terry, S. Review of 11 national policies for rare diseases in the context of key patient needs. Orphanet J. Rare Dis. 2017, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Colah, R.; Gorakshakar, A.; Nadkarni, A. Invasive & non-invasive approaches for prenatal diagnosis of haemoglobinopathies: Experiences from India. Indian J. Med. Res. 2011, 134, 552. [Google Scholar] [PubMed]
- Maryami, F.; Azarkeivan, A.; Fallah, M.S.; Zeinali, S. A Large Cohort Study of Genotype and Phenotype Correlations of Beta- Thalassemia in Iranian Population. Int. J. Hematol.-Oncol. Stem Cell Res. 2015, 9, 198–202. [Google Scholar] [PubMed]
- Hadipour Dehshal, M.; Tabrizi Namini, M.; Ahmadvand, A.; Manshadi, M.; Sadeghian Varnosfaderani, F.; Abolghasemi, H. Evaluation of the national prevention program in Iran, 2007–2009: The accomplishments and challenges with reflections on the path ahead. Hemoglobin 2014, 38, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Abolghasemi, H.; Amid, A.; Zeinali, S.; Radfar, M.H.; Eshghi, P.; Rahiminejad, M.S.; Ehsani, M.A.; Najmabadi, H.; Akbari, M.T.; Afrasiabi, A.; et al. Thalassemia in Iran: Epidemiology, prevention, and management. J. Pediatr. Hematol. Oncol. 2007, 29, 233–238. [Google Scholar] [CrossRef]
- Holtkamp, K.C.A.; Lakeman, P.; Hader, H.; Jans, S.; Hoenderdos, M.; Playfair, H.A.M.; Cornel, M.C.; Peters, M.; Henneman, L. Experiences of a High-Risk Population with Prenatal Hemoglobinopathy Carrier Screening in a Primary Care Setting: A Qualitative Study. J. Genet. Couns. 2018, 27, 635–646. [Google Scholar] [CrossRef]
- Kotila, T.R.; Adeyemo, A.A.; Mewoyeka, O.O.; Shokunb, W.A. Beta thalassaemiatriat in western Nigeria. Afr. Health Sci. 2009, 9, 46–48. [Google Scholar]
- Bento, C.; Relvas, L.; Vazão, H.; Campos, J.; Rebelo, U.; Ribeiro, M.L. The use of capillary blood samples in a large scale screening approach for the detection of beta-thalassemia and hemoglobin variants. Haematologica 2006, 91, 1565. [Google Scholar]
- Tadmouri, G.O.; Başak, A.N. β-Thalassemia in Turkey: A review of the clinical, epidemiological, molecular, and evolutionary aspects. Hemoglobin 2001, 25, 227–239. [Google Scholar] [CrossRef]
- Noori, N.; Mohamadi, M.; Keshavarz, K.; Alavi, S.M.; Mahjoubifard, M.; Mirmesdagh, Y. Comparison of right and left side heart functions in patients with thalassemia major, patients with thalassemia intermedia, and control group. J. Tehran Univ. Heart Cent. 2013, 8, 35. [Google Scholar]
- Shafie, A.A.; Chaiyakunapruk, N.; Supian, A.; Lim, J.; Zafra, M.; Hassali, M.A. State of rare disease management in Southeast Asia. Orphanet J. Rare Dis. 2016, 11, 107. [Google Scholar] [CrossRef]
- Ahmed, S.; Saleem, M.; Modell, B.; Petrou, M. Screening extended families for genetic hemoglobin disorders in Pakistan. N. Engl. J. Med. 2002, 347, 1162–1168. [Google Scholar] [CrossRef]
- Petrou, M.; Modell, B.; Shetty, S.; Khan, M.; Ward, R.H. Long-term effect of prospective detection of high genetic risk on couples’ reproductive life: Data for thalassaemia. Prenat. Diagn. 2000, 20, 469–474. [Google Scholar] [CrossRef]
- Baig, S.M.; Din, M.A.; Hassan, H.; Azhar, A.; Baig, J.M.; Aslam, M.; Anjum, I.; Farooq, M.; Hussain, M.S.; Rasool, M.; et al. Prevention of beta-thalassemia in a large Pakistani family through cascade testing. Community Genet. 2008, 11, 68–70. [Google Scholar] [CrossRef]
- Kanwal, S.; Bukhari, S.; Perveen, S. Molecular genetics and prenatal diagnosis of beta thalassemia to control transfusion dependent births in carrier Pakistani couples. J. Pak. Med. Assoc. 2017, 67, 1030–1034. [Google Scholar]
- Allen, A.; Allen, S.; Olivieri, N. Improving Laboratory and Clinical Hematology Services in Resource Limited Settings. Hematol. Oncol. Clin. North Am. 2016, 30, 497–512. [Google Scholar] [CrossRef]
- Premawardhana, A.P.; Mudiyanse, R.; De Silva, S.T.; Jiffry, N.; Nelumdeniya, U.; de Silva, U.; Lamabadusuriya, S.P.; Pushpakumara, K.; Dissanayaka, R.; Jansz, M.; et al. A nationwide survey of hospital-based thalassemia patients and standards of care and a preliminary assessment of the national prevention program in Sri Lanka. PLoS ONE 2019, 14, e0220852. [Google Scholar] [CrossRef]
- de Castro Lobo, C.L.; Ballas, S.K.; Domingos, A.C.B.; Moura, P.G.; do Nascimento, E.M.; Cardoso, G.P.; de Carvalho, S.M.F. Newborn screening program for hemoglobinopathies in Rio de Janeiro, Brazil. Pediatr. Blood Cancer 2014, 61, 34–39. [Google Scholar] [CrossRef]
- Passos-Bueno, M.R.; Bertola, D.; Horovitz, D.D.; de Faria Ferraz, V.E.; Brito, L.A. Genetics and genomics in Brazil: A promising future. Mol. Genet. Genom. Med. 2014, 2, 280–291. [Google Scholar] [CrossRef]
- Munkongdee, T.; Tanakulmas, J.; Butthep, P.; Winichagoon, P.; Main, B.; Yiannakis, M.; George, J.; Devenish, R.; Fucharoen, S.; Svasti, S. Molecular epidemiology of hemoglobinopathies in Cambodia. Hemoglobin 2016, 40, 163–167. [Google Scholar] [CrossRef]
- Hamamy, H.A.; Al-Allawi, N.A. Epidemiological profile of common haemoglobinopathies in Arab countries. J. Community Genet. 2013, 4, 147–167. [Google Scholar] [CrossRef]
- Shrestha, R.M.; Pandit, R.; Yadav, U.K.; Das, R.; Yadav, B.K.; Upreti, H.C. Distribution of Hemoglobinopathy in Nepalese population. J. Nepal. Health Res. Counc. 2020, 18, 52–58. [Google Scholar] [CrossRef]
- Mikobi, T.M.; Lukusa, P.T.; Aloni, M.N.; Lumaka, A.; Akilimali, P.Z.; Devriendt, K.; Matthijs, G.; Mbuyi Muamba, J.M.; Race, V. Association between sickle cell anemia and alpha thalassemia reveals a high prevalence of the α3. 7 triplication in congolese patients than in worldwide series. J. Clin. Lab. Anal. 2018, 32, e22186. [Google Scholar] [CrossRef]
- Mukinayi Mbiya, B.; Tumba Disashi, G.; Gulbis, B. Sickle Cell Disease in the Democratic Republic of Congo: Assessing Physicians’ Knowledge and Practices. Trop. Med. Infect. Dis. 2020, 5, 127. [Google Scholar] [CrossRef]
- Nanayakkara, K.K.; Rodrigo, U.G.; Perera, K.L.N.; Nanayakkara, C.D. Pre-natal diagnosis of thalassaemia in Sri Lanka: A ten year review. J. Obstet. Gynaecol. 2017, 37, 861–863. [Google Scholar] [CrossRef]
- Win, S. Epidemiology and Current Status of Case Management of Thalassaemia in Myanmar—Country Report. Burma Med. J. 2016, 58, 43–51. [Google Scholar]
- Fucharoen, S.; Winichagoon, P. Haemoglobinopathies in southeast Asia. Indian J. Med. Res. 2011, 134, 498–506. [Google Scholar]
- Goh, L.P.W.; Chong, E.T.J.; Lee, P.-C. Prevalence of Alpha (α)-Thalassemia in Southeast Asia (2010–2020): A Meta-Analysis Involving 83,674 Subjects. Int. J. Environ. Res. Public Health 2020, 17, 7354. [Google Scholar] [CrossRef]
- World Health Organization, Regional Office for the Eastern Mediterranean. Community Control of Genetic and Congenital Disorders; WHO: Geneva, Switzerland, 1997. [Google Scholar]
- Riewpaiboon, A.; Nuchprayoon, I.; Torcharus, K.; Indaratna, K.; Thavorncharoensap, M.; Ubol, B.O. Economic burden of beta-thalassemia/Hb E and beta-thalassemia major in Thai children. BMC Res. Notes 2010, 3, 29. [Google Scholar] [CrossRef]
- Karnon, J.; Zeuner, D.; Brown, J.; Ades, A.E.; Wonke, B.; Modell, B. Lifetime treatment costs of beta-thalassaemia major. Clin. Lab. Haematol. 1999, 21, 377–385. [Google Scholar] [CrossRef]
- Ginsberg, G.; Tulchinsky, T.; Filon, D.; Goldfarb, A.; Abramov, L.; Rachmilevitz, E.A. Cost-benefit analysis of a national thalassaemia prevention programme in Israel. J. Med. Screen. 1998, 5, 120–126. [Google Scholar] [CrossRef] [PubMed]
- de Silva, S.; Fisher, C.A.; Premawardhena, A.; Lamabadusuriya, S.P.; Peto, T.E.; Perera, G.; Old, J.M.; Clegg, J.B.; Olivieri, N.F.; Weatherall, D.J. Thalassaemia in Sri Lanka: Implications for the future health burden of Asian populations. Sri Lanka Thalassaemia Study Group. Lancet 2000, 355, 786–791. [Google Scholar] [CrossRef]
- Aung Myo, H.; Khin Ei, H.; Thein Thein, M. Thalassemia in the outpatient department of the Yangon Children’s Hospital in Myanmar: Cost analysis of the day-care-room services for thalassemia. Southeast Asian J. Trop. Med. Public Health 1992, 23, 273–277. [Google Scholar]
- Dhamcharee, V.; Romyanan, O.; Ninlagarn, T. Genetic counseling for thalassemia in Thailand: Problems and solutions. Southeast Asian J. Trop. Med. Public Health 2001, 32, 413–418. [Google Scholar]
- Cao, A.; Kan, Y.W. The prevention of thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011775. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.S.; Phak, N.H.; Ying, T.P.; Choo, H.L.; Muda, Z.; Bahrin, S.; Kaur, B.; Jamal, R.; Keong, T.M.; George, E. Management of Thalassaemia. 2003. Available online: https://www.moh.gov.my/moh/resources/auto%20download%20images/587f136ce4807.pdf (accessed on 3 January 2022).
- Atkin, K.; Ahmad, W.I. Living a ‘normal’ life: Young people coping with thalassaemia major or sickle cell disorder. Soc. Sci. Med. 2001, 53, 615–626. [Google Scholar] [CrossRef]
- Ahmed, S.; Green, J.M.; Hewison, J. Attitudes towards prenatal diagnosis and termination of pregnancy for thalassaemia in pregnant Pakistani women in the North of England. Prenat. Diagn. 2006, 26, 248–257. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, G. Problems of screening and counseling in the hemoglobinopathies. Birth Defects Amst. Excerpta Med. 1974, 268–276. [Google Scholar]
- Stevens, G.A.; Alkema, L.; Black, R.E.; Boerma, J.T.; Collins, G.S.; Ezzati, M.; Grove, J.T.; Hogan, D.R.; Hogan, M.C.; Horton, R.; et al. Guidelines for Accurate and Transparent Health Estimates Reporting: The GATHER statement. Lancet 2016, 388, e19–e23. [Google Scholar] [CrossRef]


| Qualitative Variables in ITHANET |
| Major Qualitative Variables (Availability of the following parameters) |
| 1. Genetic counselling |
| 2. Haemoglobinopathies patient registry |
| 3. Dedicated treatment centres |
| 4. Blood transfusion availability |
| 5. Iron chelation availability |
| 6. Prevention programme |
| 7. Prenatal screening |
| 8. Antenatal screening |
| 9. Patient associations |
| Minor Qualitative Variables (Availability of the following parameters) |
| 1. MRI facilities |
| 2. SCD or thalassaemia newborn screening |
| Quantitative Variables in ITHANET |
| 1. Prevalence of β-thalassaemia carriers |
| 2. Prevalence of α-thalassaemia carriers |
| 3. Expected incidence of β-thalassaemia |
| 4. Mutation frequencies |
| 5. Known sickle cell disease patients |
| 6. Prevalence of sickle cell disease carriers |
| 7. Incidence of sickle cell disease |
| 8. Prevalence of Hb E carriers |
| 9. Prevalence of Hb C carriers |
| 10. Known β-thalassaemia patients |
| 11. Incidence of β-thalassaemia |
| Category | Short Description | Criterion for Group Scoring | GGN Member Countries | Accumulative Scoring * |
|---|---|---|---|---|
| A | Countries where services are well-established with a national system for prevention and control | All 9 major qualitative variables are present | Cyprus | A0 |
| France | A1,2 | |||
| Italy | A1,2 | |||
| Malaysia | A1 | |||
| Singapore | A0 | |||
| United Kingdom (UK) | A1,2 | |||
| B | Countries with efforts to create a partial/fragmented national control programme in place, but with limited availability/accessibility | 1–3 major qualitative variables are absent or data are not available | Australia | B1 |
| Bangladesh | B0 | |||
| Belgium | B2 | |||
| China | B0 | |||
| India | B2 | |||
| Indonesia | B1 | |||
| Iran | B0 | |||
| Netherland | B1,2 | |||
| Nigeria | B0 | |||
| Pakistan | B0 | |||
| Portugal | B1 | |||
| South Africa | B1 | |||
| Spain | B2 | |||
| Thailand | B1 | |||
| Turkey | B1 | |||
| Vietnam | B0 | |||
| C | Countries where expertise on haemoglobinopathy data collection and management exists but is not part of a sustainable national control programme | 4–6 major qualitative variables are absent or data are not available | Brazil | C1 |
| Cambodia | C0 | |||
| Egypt | C0 | |||
| Nepal | C0 | |||
| Philippines | C2 | |||
| Dem. Rep. of the Congo | C1,2 | |||
| Sri Lanka | C0 | |||
| D | Countries where expertise and infrastructure for haemoglobinopathy data collection and management are limited | >6 major qualitative variables are absent or data are not available | Brunei | D0 |
| Laos | D0 | |||
| Myanmar | D0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halim-Fikri, B.H.; Lederer, C.W.; Baig, A.A.; Mat-Ghani, S.N.A.; Syed-Hassan, S.-N.R.-K.; Yusof, W.; Abdul Rashid, D.; Azman, N.F.; Fucharoen, S.; Panigoro, R.; et al. Global Globin Network Consensus Paper: Classification and Stratified Roadmaps for Improved Thalassaemia Care and Prevention in 32 Countries. J. Pers. Med. 2022, 12, 552. https://doi.org/10.3390/jpm12040552
Halim-Fikri BH, Lederer CW, Baig AA, Mat-Ghani SNA, Syed-Hassan S-NR-K, Yusof W, Abdul Rashid D, Azman NF, Fucharoen S, Panigoro R, et al. Global Globin Network Consensus Paper: Classification and Stratified Roadmaps for Improved Thalassaemia Care and Prevention in 32 Countries. Journal of Personalized Medicine. 2022; 12(4):552. https://doi.org/10.3390/jpm12040552
Chicago/Turabian StyleHalim-Fikri, Bin Hashim, Carsten W. Lederer, Atif Amin Baig, Siti Nor Assyuhada Mat-Ghani, Sharifah-Nany Rahayu-Karmilla Syed-Hassan, Wardah Yusof, Diana Abdul Rashid, Nurul Fatihah Azman, Suthat Fucharoen, Ramdan Panigoro, and et al. 2022. "Global Globin Network Consensus Paper: Classification and Stratified Roadmaps for Improved Thalassaemia Care and Prevention in 32 Countries" Journal of Personalized Medicine 12, no. 4: 552. https://doi.org/10.3390/jpm12040552
APA StyleHalim-Fikri, B. H., Lederer, C. W., Baig, A. A., Mat-Ghani, S. N. A., Syed-Hassan, S.-N. R.-K., Yusof, W., Abdul Rashid, D., Azman, N. F., Fucharoen, S., Panigoro, R., Silao, C. L. T., Viprakasit, V., Jalil, N., Mohd Yasin, N., Bahar, R., Selvaratnam, V., Mohamad, N., Nik Hassan, N. N., Esa, E., ... on behalf of the Global Globin Network (GGN). (2022). Global Globin Network Consensus Paper: Classification and Stratified Roadmaps for Improved Thalassaemia Care and Prevention in 32 Countries. Journal of Personalized Medicine, 12(4), 552. https://doi.org/10.3390/jpm12040552