
COL-3-Induced Molecular and Ultrastructural Alterations in K562 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells Lines and Cell Culture
2.2. Cell Treatment
2.3. Measurement of Cytotoxicity
2.4. Cell Death Assessment
2.5. Transmission Electron Microscopy (TEM)
2.6. Protein Analysis Using Western Blotting
2.7. Intracellular Reactive Oxygen Species Assay
2.8. Immunocytochemistry (ICC)
2.9. Statistical Analysis
3. Results
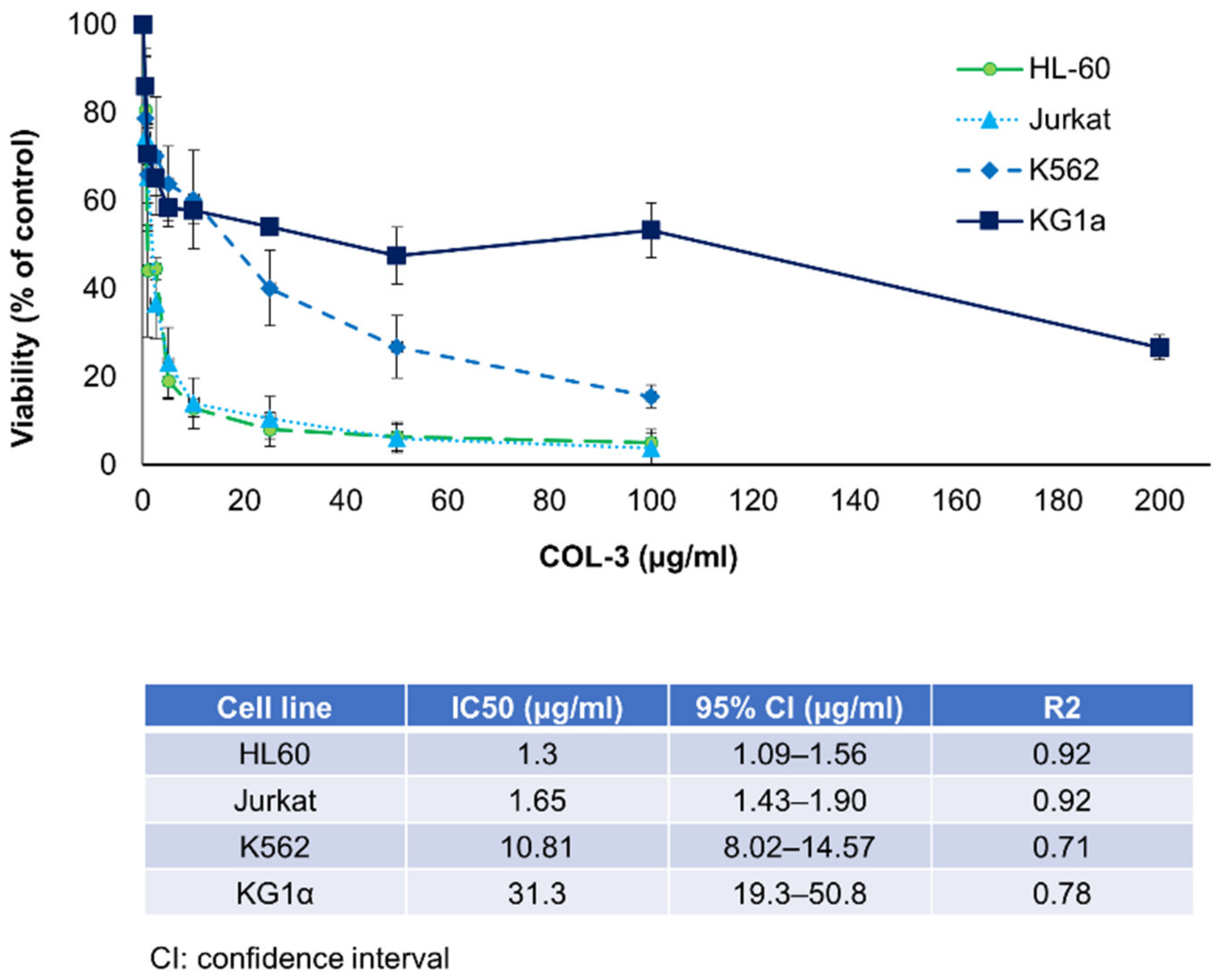
3.1. Effects of COL-3 on the Viability of K562 Cells
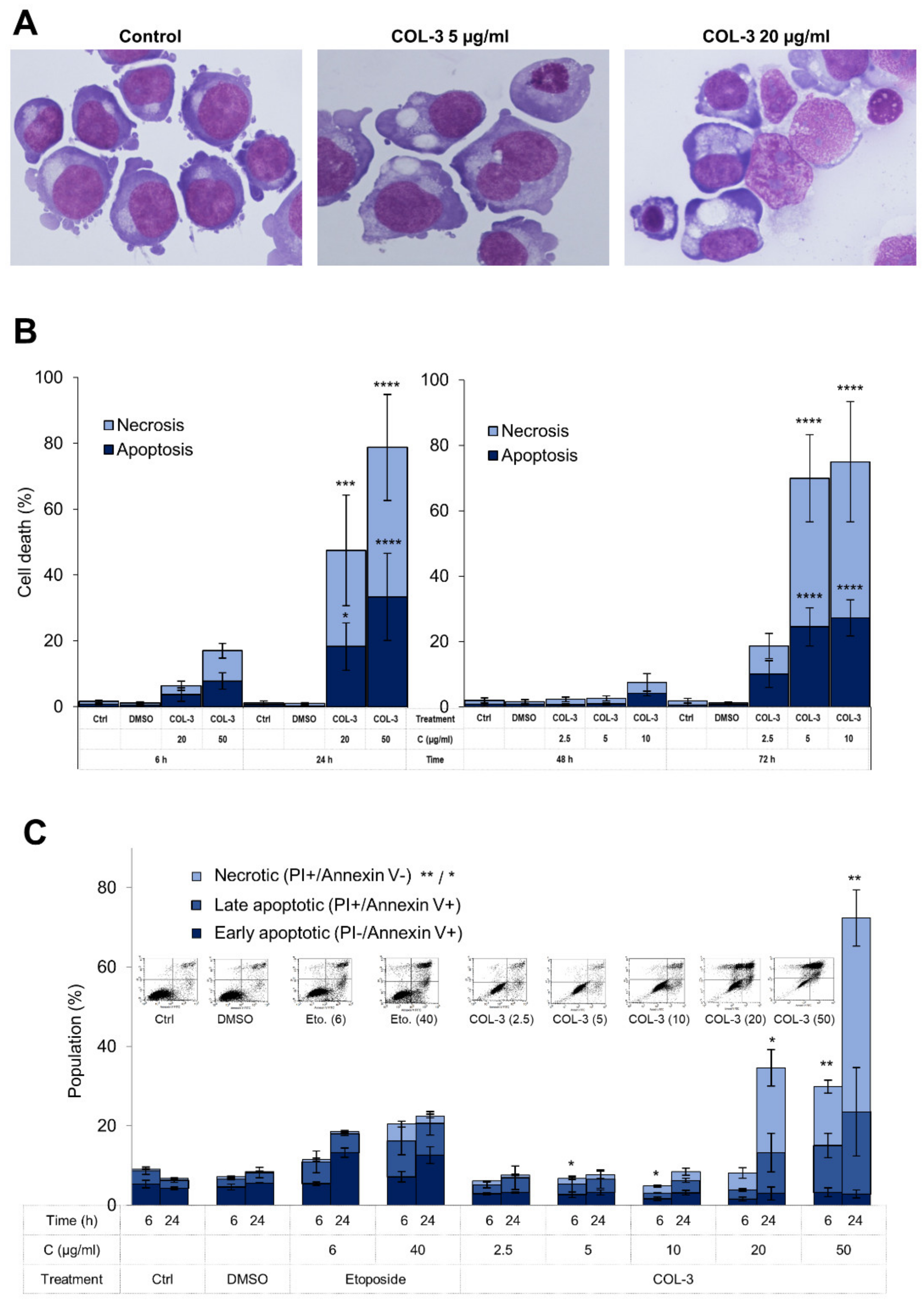
3.2. Characteristics of COL-3-Induced Cell Death in K562 Cells
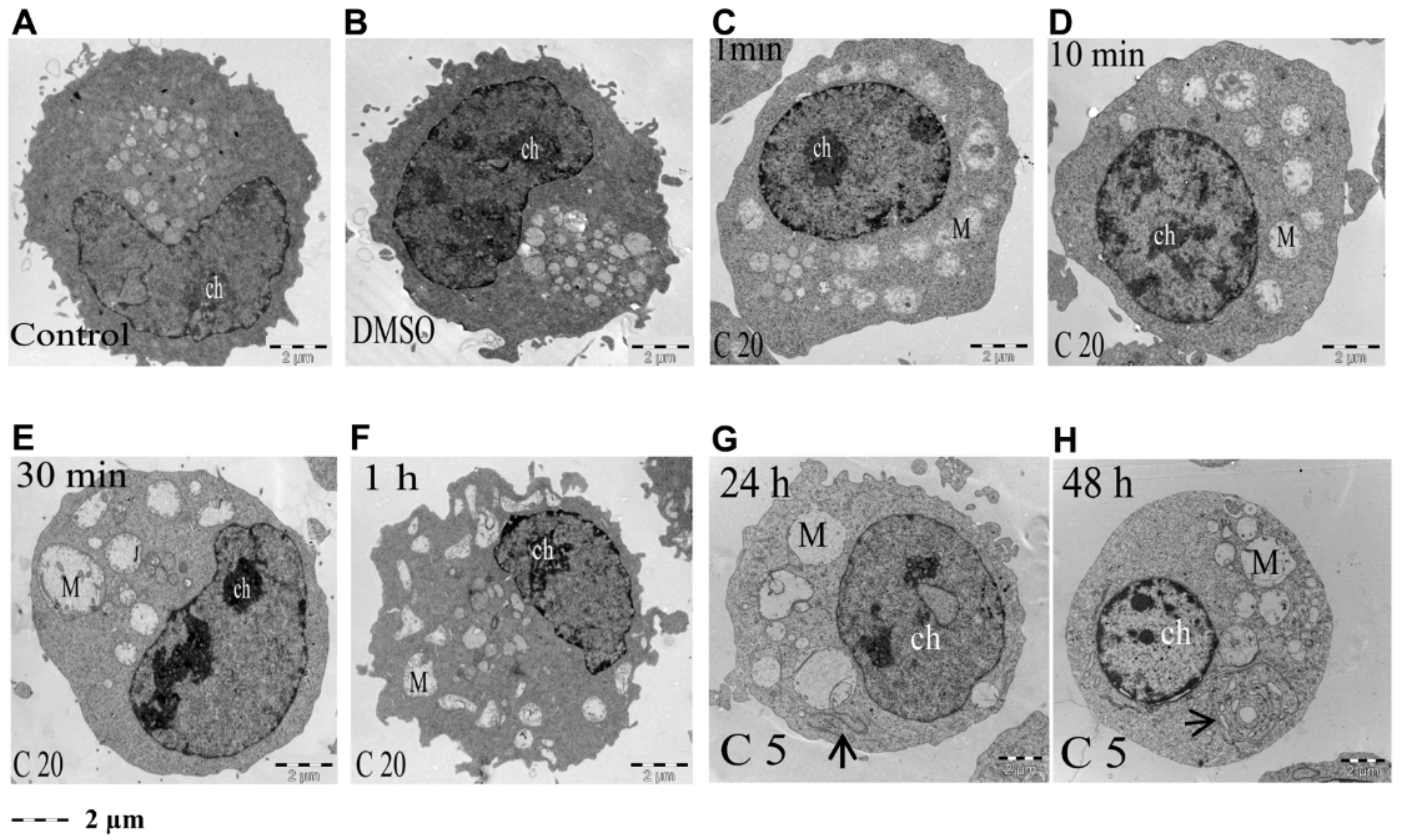
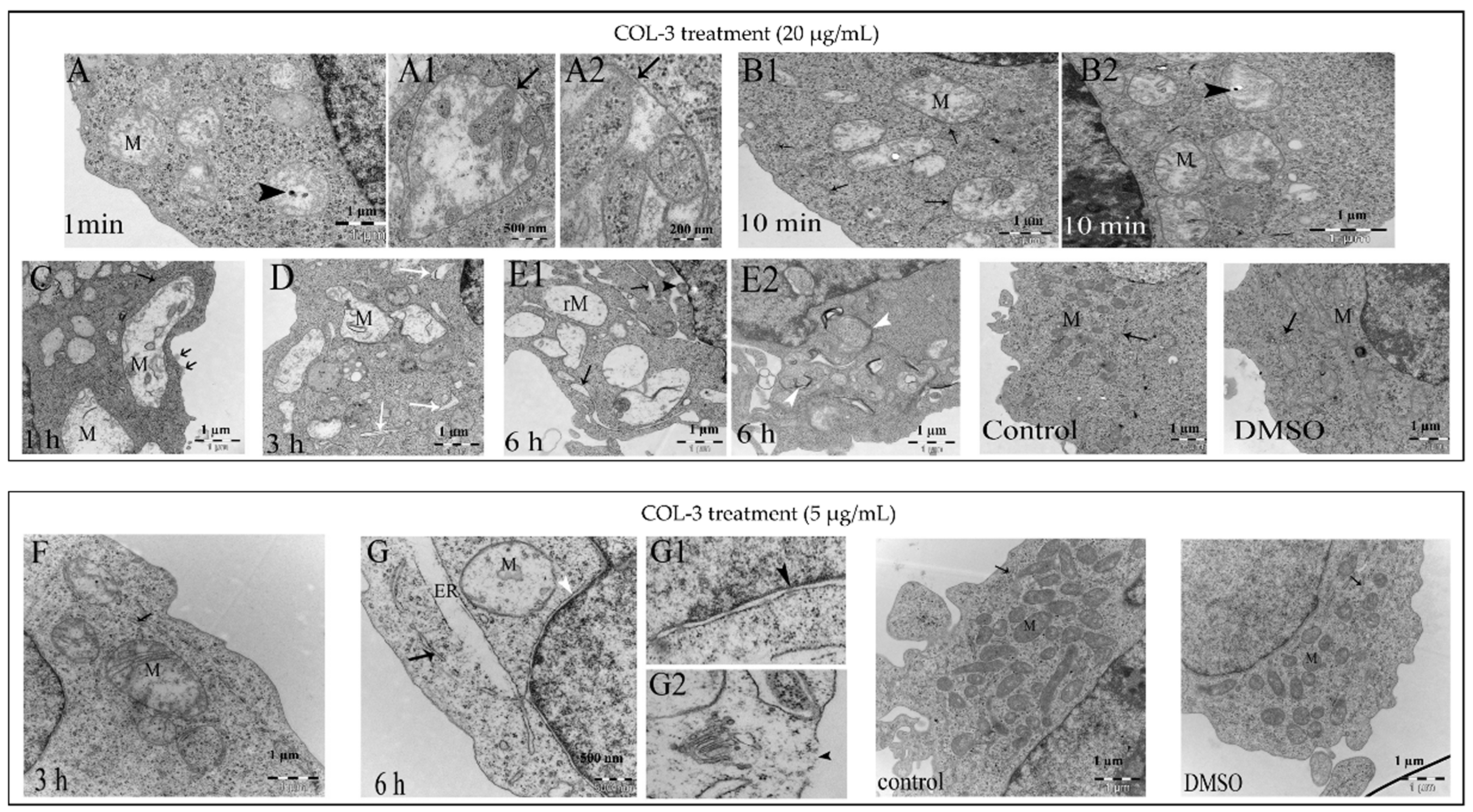
3.3. Induction of Ultrastructural Changes in K562 Cells by COL-3
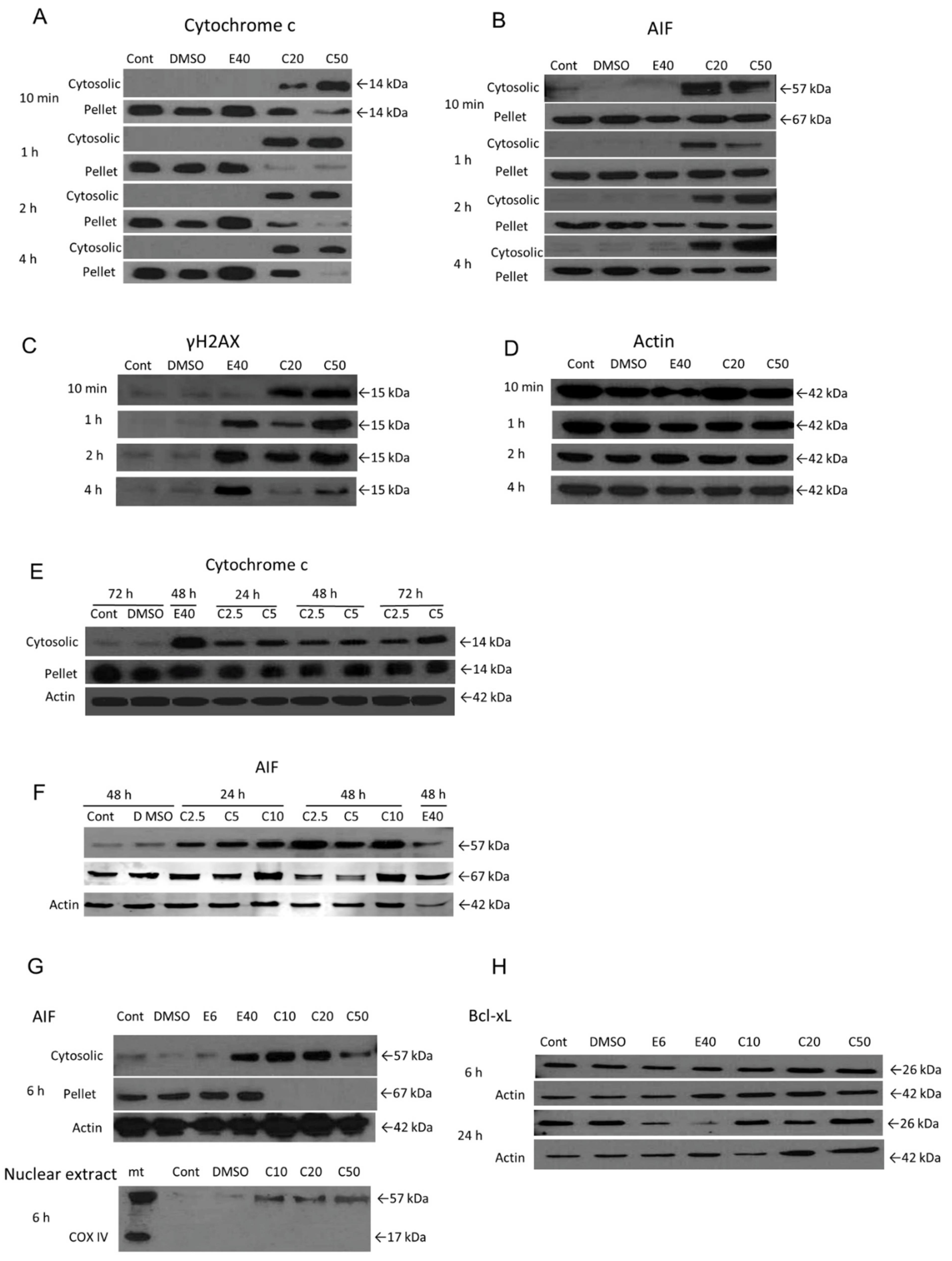
3.4. The Role of Mitochondrial Mediators of Apoptosis in COL-3-Induced Cell Death in K562 Cells
3.5. COL-3 Induced DNA Damage in K562 Cells
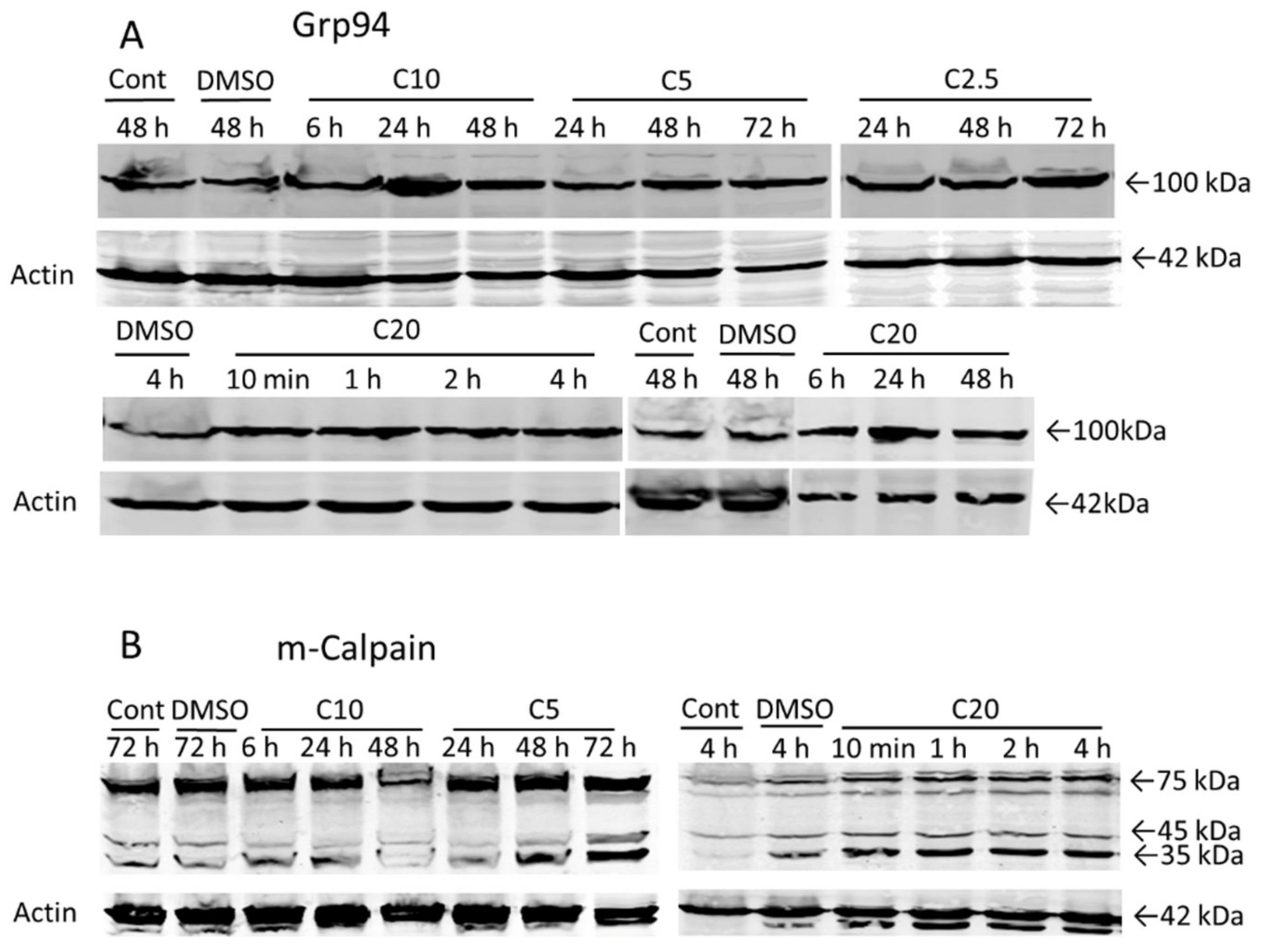
3.6. Relationship between COL-3-Induced Dilatation and the Development of Cell Death
3.7. The Role of Caspase Activation and ROS Formation in COL-3 Induced Cell Death
3.8. Effect of Extracellular Ion Concentrations on COL-3-Induced Toxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Leverkus, M. Programmed necrosis and necroptosis signalling. FEBS J. 2015, 282, 19–31. [Google Scholar] [CrossRef]
- Nelson, M.L. Chemical and biological dynamics of tetracyclines. Adv. Dent. Res. 1998, 12, 5–11. [Google Scholar] [CrossRef]
- Khan, M.A.; Musarrat, J. Interactions of tetracycline and its derivatives with DNA in vitro in presence of metal ions. Int. J. Biol. Macromol. 2003, 33, 49–56. [Google Scholar] [CrossRef]
- Pinsuwan, S.; Alvarez-Nunez, F.A.; Tabibi, S.E.; Yalkowsky, S.H. Spectrophotometric determination of acidity constants of 4-dedimethylamino sancycline (Col-3), a new antitumor drug. J. Pharm. Sci. 1999, 88, 535–537. [Google Scholar] [CrossRef]
- Lokeshwar, B.L.; Selzer, M.G.; Zhu, B.Q.; Block, N.L.; Golub, L.M. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int. J. Cancer 2002, 98, 297–309. [Google Scholar] [CrossRef]
- Onoda, T.; Ono, T.; Dhar, D.K.; Yamanoi, A.; Nagasue, N. Tetracycline analogues (doxycycline and COL-3) induce caspase-dependent and -independent apoptosis in human colon cancer cells. Int. J. Cancer 2006, 118, 1309–1315. [Google Scholar] [CrossRef]
- Song, H.; Fares, M.; Maguire, K.R.; Siden, A.; Potacova, Z. Cytotoxic effects of tetracycline analogues (doxycycline, minocycline and COL-3) in acute myeloid leukemia HL-60 cells. PLoS ONE 2014, 9, e114457. [Google Scholar] [CrossRef] [Green Version]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Nestal de Moraes, G.; Souza, P.S.; Costas, F.C.; Vasconcelos, F.C.; Reis, F.R.; Maia, R.C. The Interface between BCR-ABL-Dependent and -Independent Resistance Signaling Pathways in Chronic Myeloid Leukemia. Leuk. Res. Treat. 2012, 2012, 671702. [Google Scholar] [CrossRef] [Green Version]
- Durland-Busbice, S.; Reisman, D. Lack of p53 expression in human myeloid leukemias is not due to mutations in transcriptional regulatory regions of the gene. Leukemia 2002, 16, 2165–2167. [Google Scholar] [CrossRef] [Green Version]
- Daugas, E.; Nochy, D.; Ravagnan, L.; Loeffler, M.; Susin, S.A.; Zamzami, N.; Kroemer, G. Apoptosis-inducing factor (AIF): A ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000, 476, 118–123. [Google Scholar] [CrossRef]
- Wynand, P.R.; Bernd, K. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signaling—from basic mechanisms to clinical application. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Steven, M.H.; Muhammad, M.Y.; David, A. Allen Caspase and calpain function in cell death: Bridging the gap between apoptosis and necrosis. Ann. Clin. Biochem. 2005, 42 Pt 6, 415–431. [Google Scholar]
- Peterson, L.F.; Mitrikeska, E.; Giannola Lui Sun, H.; Bixby, D.; Malek, S.N.; Donato, N.; Wang, S.; Talpaz, M. p53 stabilization induces apoptosis in chronic myeloid leukemia blast crisis cells. Leukemia 2011, 25, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Cioe, L.; McNab, A.; Hubbell, H.R.; Meo, P.; Curtis, P.; Rovera, G. Differential expression of the globin genes in human leukemia K562(S) cells induced to differentiate by hemin or butyric acid. Cancer Res. 1981, 41, 237–243. [Google Scholar]
- Palade, G. Intracellular aspects of the process of protein synthesis. Science 1975, 189, 867. [Google Scholar] [CrossRef]
- Mortison, J.D.; Schenone, M.; Myers, J.A.; Zhang, Z.; Chen, L.; Ciarlo, C.; Comer, E.; Natchiar, S.K.; Carr, S.A.; Klaholz, B.P.; et al. Tetracyclines Modify Translation by Targeting Key Human rRNA Substructures. Cell Chem. Biol. 2018, 25, 1506–1518.e13. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; McStay, G.P.; Clarke, S.J. The permeability transition pore complex: Another view. Biochimie 2002, 84, 153–166. [Google Scholar] [CrossRef]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Zamzami, N.; Larochette, N.; Kroemer, G. Mitochondrial permeability transition in apoptosis and necrosis. Cell Death Differ. 2005, 12 (Suppl. 2), 1478–1480. [Google Scholar] [CrossRef]
- Ozaki, T.; Yamashita, T.; Ishiguro, S. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim. Biophys. Acta 2009, 1793, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.J.; Guay, G.; Pogan, L.; Sauve, R.; Nabi, I.R. Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J. Cell. Biol. 2000, 150, 1489–1498. [Google Scholar] [CrossRef]
- Gutierrez-Diaz, J.A.; Cuevas, P.; Reimers, D.; Dujovny, M.; Diaz, F.G.; Ausman, J.I. Quantitative electron microscopic study of calcium accumulation in cerebral ischemia mitochondria. Surg. Neurol. 1985, 24, 67–72. [Google Scholar] [CrossRef]
- Pizzo, P.; Scapin, C.; Vitadello, M.; Florean, C.; Gorza, L. Grp94 acts as a mediator of curcumin-induced antioxidant defence in myogenic cells. J. Cell. Mol. Med. 2010, 14, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Gurfinkel, D.M.; Chow, S.; Hurren, R.; Gronda, M.; Henderson, C.; Berube, C.; Hedley, D.W.; Schimmer, A.D. Disruption of the endoplasmic reticulum and increases in cytoplasmic calcium are early events in cell death induced by the natural triterpenoid Asiatic acid. Apoptosis 2006, 11, 1463–1471. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone Deacetylase Regulation of ATM-Mediated DNA Damage Signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Boujrad, H.; Gubkina, O.; Robert, N.; Krantic, S.; Susin, S.A. AIF-mediated programmed necrosis: A highly regulated way to die. Cell Cycle 2007, 6, 2612–2619. [Google Scholar] [CrossRef] [Green Version]
- Georgina, G.A.; Bettina, S.; Daniel, M.; Carlos, R.A.; Armando, G.H.; Ramces, F.V. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar]
- Mary, C.; Timothy, P.C.; Jeannette, Y.L.; Michelle, A.R.; David, T.S.; Lee, R.; James, M.P.; William, D.F.; Susan, E.K.; Bruce, J.D. Matrix Metalloproteinase Inhibitor COL-3 in the Treatment of AIDS-Related Kaposi’s Sarcoma: A Phase I AIDS Malignancy Consortium Study. J. Clin. Oncol. 2002, 20, 153–159. [Google Scholar]
- Bruce, J.D.; Susan, E.K.; Jeannette, Y.L.; Kenneth, S.B.; David, M.A. Randomized Phase II Trial of Matrix Metalloproteinase Inhibitor COL-3 in AIDS-Related Kaposi’s Sarcoma: An AIDS Malignancy Consortium Study. J. Clin. Oncol. 2006, 24, 1389–1394. [Google Scholar]
- Fumio, N.; Jiro, K.; Tomoyuki, U.; Chiemi, N.; Katsuhiro, T.; Daichi, I.; Toshihiro, M.; Yuki, K.; Yutaka, E.; Kimihito, C.K.; et al. Hes1 promotes blast crisis in chronic myelogenous leukemia through MMP-9 upregulation in leukemic cells. Blood 2014, 123, 3932–3942. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fares, M.; Oerther, S.; Hultenby, K.; Gubrianska, D.; Zhao, Y.; Abedi-Valugerdi, M.; Hassan, M. COL-3-Induced Molecular and Ultrastructural Alterations in K562 Cells. J. Pers. Med. 2022, 12, 42. https://doi.org/10.3390/jpm12010042
Fares M, Oerther S, Hultenby K, Gubrianska D, Zhao Y, Abedi-Valugerdi M, Hassan M. COL-3-Induced Molecular and Ultrastructural Alterations in K562 Cells. Journal of Personalized Medicine. 2022; 12(1):42. https://doi.org/10.3390/jpm12010042
Chicago/Turabian StyleFares, Mona, Sandra Oerther, Kjell Hultenby, Danica Gubrianska, Ying Zhao, Manuchehr Abedi-Valugerdi, and Moustapha Hassan. 2022. "COL-3-Induced Molecular and Ultrastructural Alterations in K562 Cells" Journal of Personalized Medicine 12, no. 1: 42. https://doi.org/10.3390/jpm12010042
APA StyleFares, M., Oerther, S., Hultenby, K., Gubrianska, D., Zhao, Y., Abedi-Valugerdi, M., & Hassan, M. (2022). COL-3-Induced Molecular and Ultrastructural Alterations in K562 Cells. Journal of Personalized Medicine, 12(1), 42. https://doi.org/10.3390/jpm12010042