Abstract
Galactosemia is a rare inherited metabolic disease resulting from mutations in the four genes which encode enzymes involved in the metabolism of galactose. The current therapy, the removal of galactose from the diet, is inadequate. Consequently, many patients suffer lifelong physical and cognitive disability. The phenotype varies from almost asymptomatic to life-threatening disability. The fundamental biochemical cause of the disease is a decrease in enzymatic activity due to failure of the affected protein to fold and/or function correctly. Many novel therapies have been proposed for the treatment of galactosemia. Often, these are designed to treat the symptoms and not the fundamental cause. Pharmacological chaperones (PC) (small molecules which correct the folding of misfolded proteins) represent an exciting potential therapy for galactosemia. In theory, they would restore enzyme function, thus preventing downstream pathological consequences. In practice, no PCs have been identified for potential application in galactosemia. Here, we review the biochemical basis of the disease, identify opportunities for the application of PCs and describe how these might be discovered. We will conclude by considering some of the clinical issues which will affect the future use of PCs in the treatment of galactosemia.
1. Introduction
Galactosemia is an inherited metabolic disease which causes deficiency in the metabolism of galactose and in the formation of galactose containing products in the body [1,2,3,4,5,6]. In normal metabolism, the Leloir pathway takes galactose and converts it into glucose 1-phosphate (Figure 1). This product can be further metabolized to glucose 6-phosphate, which enters glycolysis for energy production in the cell. The Leloir pathway has four steps, each catalysed by a different enzyme [7,8,9]. The types of galactosemia are segregated depending on which enzyme the deficiency is present in [2].
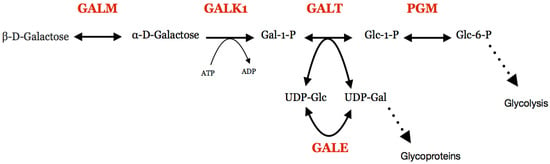
Figure 1.
The Leloir pathway and associated reactions. Protein names are shown in red. GALM, galactose mutarotase; GALK1, galactokinase; GALT, galactose 1-phosphate uridylyltransferase; PGM, Phosphoglucomutase; GALE, UDP -galactose 4′-epimerase; Gal, galactose; Glc, glucose.
Clinical manifestations of the disease include cataracts, low IQ, neonatal jaundice, infantile failure to thrive, Escherichia coli sepsis, neuromuscular dysfunction, infertility or miscarriage [10,11,12,13,14]. Most severe symptoms occur in early life when children are dependent on milk for nutrition. The severity of symptoms depends on which enzyme is affected, the mutation’s effect on the protein and the patient’s environment, including diet. Some of the effects of the condition can be prevented by the avoidance of galactose containing foods in the diet, which should start as soon as possible after birth [11,15].
Type I galactosemia (OMIM#230400), also known as classic galactosemia, is the most commonly diagnosed, and often has the most severe symptoms. It is caused by a mutation in the GALT gene which codes for the enzyme galactose 1-phosphate uridylyltransferase (GALT; EC 2.7.7.12) [16,17]. p.Q188R is the most common mutation in Caucasians, manifesting with life-threatening symptoms in the neonatal period [18,19]. In the black population, the p.S135L variant is more commonly detected. Phenotypically, it is associated with slightly better clinical outcomes than p.Q188R [20,21,22]. Type II galactosemia (previously known as galactokinase deficiency; OMIM#230200) is caused by mutations in the gene coding for the enzyme galactokinase (GALK1; EC 2.7.1.6) [23,24]. It is rarer than type I, and often less severe, typically manifesting in early onset cataracts [25]. Type III galactosemia (OMIM#230350) is caused by mutations in the GALE gene, which codes for the enzyme UDP-galactose 4′-epimerase (GALE; EC 5.1.3.2) [26]. It usually has the mildest symptoms. In some forms, it is thought the disease only affects red blood cells. The most severe forms are phenotypically similar to type I galactosemia [27,28]. This enzyme is not only critical for the use of galactose as an energy source, but also in the process of attaching galactose sidechains to other proteins, carbohydrates and lipids [29]. Type IV galactosemia (OMIM#137030) is caused by mutations in the GALM gene causing deficiency in the galactose mutarotase enzyme (GALM; EC 5.1.3.3) [30,31,32,33]. It has only been recently discovered that mutations in this gene lead to a disease state. It presents with symptoms similar to type II [31].
The most commonly used diagnostic test for galactosemia is the Beulter test (Figure 2) [34]. This relies on detecting the conversion of galactose 1-phosphate to gluconate 6-phosphate, a process which requires the activities of GALT, phosphoglucomutase (PGM) and glucose 6-phosphate dehydrogenase (G6PDH). The last step uses NADP+ as an oxidizing agent. The reduced cofactor, NADPH, can be detected by fluorescence. Thus, a positive result for type I galactosemia is the absence of this fluorescence. False positives result from blood samples which contain EDTA, samples exposed to heat, and deficiency of PGM or G6PDH [35]. An alternative test uses a non-specific phosphatase enzyme to convert galactose 1-phosphate to galactose. Galactose is then detected by oxidation and catalysed by galactose dehydrogenase, an enzyme which does not occur in humans. This requires NAD+ as a cofactor, and the presence of the reduced form indicates a positive result and is detected by fluorescence [36]. Tandem mass spectrometry is also a widely used method to determine both galactose 1-phosphate [37] concentration and GALT activity [38]. Galactose 1-phosphate concentrations are raised in the blood of type I (and sometimes type III) galactosemia patients. While concentrations of this metabolite are not raised in type II galactosemia, galactose concentrations are increased and so false positives and consequent misdiagnosis can occur. Diagnosis is normally confirmed by molecular biology methods. Historically, these methods involved the use of site-specific probes to identify common mutations, which inevitably missed rarer and novel mutations. Therefore, complete sequencing of the suspected causative gene is recommended to ensure a personalized medicine approach. Some countries include galactosemia in newborn screening programs [39]. In many cases, only GALT deficiency is tested for since this is the most common and severe form, but lower rates of testing for the other types could be one reason why they are less commonly diagnosed. It is also possible that types II, III and IV can result in very mild or almost asymptomatic phenotypes which remain undiagnosed. For example, the p.A198V variant of GALK1 results in an increased risk of cataracts in mid and later life [40]. A further useful test is measuring urine galactitol, which can be performed by GC/MS or gas chromatography [41]. Enzyme levels can also be determined through enzymatic assays.
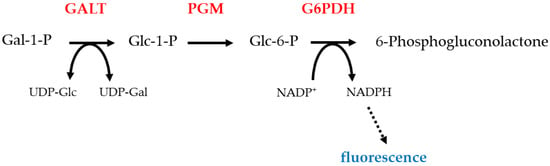
Figure 2.
Beulter test: this test detects the fluorescence produced by the NADPH. NADPH is made by the action of galactose 6-phosphate dehydrogenase on glucose 6-phosphate [34]. The absence of fluorescence, which would occur in GALT deficiency, is a positive test. Protein names are shown in redGALT, galactose 1-phosphate uridylyltransferase; PGM, Phosphoglucomutase; G6PDH, glucose-6-phosphate dehydrogenase; Gal, galactose; Glc, glucose.
Attempts have been made to try and elucidate how the genotype of galactosemia links to the phenotype expressed in patients by using yeast, mouse, rat, nematode, zebra fish and fly models [42,43,44,45,46,47,48,49,50,51,52,53,54]. However, significant questions are still unanswered. Mechanisms such as the build-up of toxic metabolites (i.e., galactose, galactose 1-phosphate or galactitol) have been thought to explain some but not all symptoms [55]. How these increased metabolite levels result in complex pathology affecting multiple organ systems is contentious.
Apart from the avoidance of galactose containing food products, no other specific treatments are commonly used in clinical practice. Other treatments used are symptom specific, such as fertility treatments for female patients [14]. The total elimination of galactose is impossible as some galactose is made in vivo, and some is required in the making of galactose containing products (in type III only). Therefore, long-term side-effects are inevitable [56,57,58]. Treatments under investigation include GALK1 inhibitors, antioxidants, aldose reductase inhibitors, gene therapy, mRNA therapy, stress signalling pathway inhibitors and enzyme replacement therapy (recently reviewed in [59,60,61,62,63]). Another potential strategy is the use of pharmacological chaperones (PCs). This strategy works by using a small molecule to bind to the affected protein to increase its stability, as many of the disease-causing mutations cause protein instability [64,65,66,67]. The chaperone may also aid in preventing protein misfolding (see below).
2. Pharmacological Chaperones
PCs are a potential therapeutic approach for protein misfolding and aggregation diseases [68]. PCs consist of small molecules that bind to the target proteins, stabilising disease-associated variants [69,70]. Their preferential binding to the native/folded state prevents the conformational fluctuations from destabilising the target protein, hindering misfolding and aggregation. This then shifts the equilibrium towards the folded form, resulting in a higher amount of protein that avoids the quality control machinery, decreases aberrant trafficking, and ultimately increases the active pool of enzyme. As such, PCs have been proposed to correct the folding and trafficking defects for not only enzymes, but transporters, receptors, and other structural proteins as well [68]. Like other small molecule drugs, PCs have a number of potential advantages over other treatments, such as oral delivery, broad biodistribution, and lower costs. They can be less burdensome to patients. Importantly, their ability to cross the blood–brain barrier to reach different target tissues can allow for the treatment of neuropathic pathologies present in many rare disorders. It has been proposed that increasing protein function to moderate levels beyond a threshold (e.g., 10%) would be sufficient to delay the onset and slow the clinical progression of these disorders [68]. Notably, though suggested to target stability and misfolding issues of disease-associated variants, PCs can likewise stabilise and increase levels of the wild type protein, which can increase the efficacy of enzyme replacement therapies (ERT) if co-administered [71]. For a small molecule to act as a PC, it must have a (reasonably) high binding affinity and specificity for the wild type or disease variant of the protein, higher in the folded than in partially folded state(s). The binding affinity of the PC molecule generally correlates with its potential to rescue misfolding and aggregation. Fundamentally, native ligands of target enzymes, such as substrates, cofactors, and products with well-known interactions, can serve as templates for the development of PCs [72]. Therefore, early-stage proof-of-concept and discovery studies have relied heavily upon protein structural information, allowing for in-depth analysis at the molecular level of the effects of disease associated mutations and characterising protein-small molecule interactions for drug design.
For misfolding disorders, a number of PC therapies have reached the clinical setting, each with their own specific mode of action serving as paradigms for efforts in other disorders. An example of a substrate mimetic PC can be found in the development for the treatment of Fabry disease (OMIM #301500), an X-linked disorder that reduces the activity of α-galactosidase A (GLA). This disease is representative of lysosomal storage disorders (LSDs) [73]. LSDs are a group of inherited diseases characterized by toxic accumulation of glycosphingolipids, glycoproteins or mucopolysaccharides in patient lysosomes. The iminosugar 1-deoxygalactonojirimycin (DGJ, migalastat hydrochloride) mimics the terminal galactose of GLA’s native substrate and has been shown to increase GLA activity in cultured cells, mouse models, and clinical trials [74], leading to the approval of the drug by the European Union in 2016 (Galafold) [74].
Another example is cystic fibrosis (CF, OMIM #219700). CF is caused by defects in the ATP-binding cassette -type chloride channel CFTR (cystic fibrosis transmembrane regulator). Commonly, mutations result in protein misfolding and mis-trafficking, which leads to proteasome degradation [75]. Consequently, there have been intensive efforts aimed at developing PCs that either increase channel activity (“potentiator”) or improve CFTR trafficking (“corrector”) [76]. These efforts have resulted in the development of Ivacaftor, an FDA-approved potentiator. Invacaftor increases channel activity and lung function in CF patients with the p.G551D variant. Lumacaftor/Tezacaftor are correctors that improve the folding and increase trafficking of the p.ΔF508 variant, which is the most common mutation for CF [77].
A PC therapy for hereditary transthyretin (TTR)-related amyloidosis (OMIM #105210) has also been developed. This disorder is an autosomal dominant disease of progressive neuropathies and cardiopathies caused by mutations in TTR. TTR is a transporter for thyroxine and holo retinol-binding protein. Disease-related mutations tend to cause TTR destabilization, leading to the native tetramer disassociating into monomers that misfold and trigger amyloid aggregation [78]. Therefore, a PC approach that stabilizes the tetrameric form, reducing the build-up of the amyloidogenic monomer and preventing TTR aggregation, was developed: Tafamidis, a benzoxazole derivative which has been shown to slow disease progression for certain genotypes of the disorder [79]. Significantly, the crystal structure reveals how this molecule occupies the two thyroxine-binding sites that are usually unoccupied under physiological conditions [80]. Binding causes the stabilization of the dimer–dimer interface and provides a structural basis for kinetic stabilization. This reduces the tetramer-to-monomer disassociation and thus prevents amyloidogenesis.
3. Potential in the Treatment of Galactosemia
The fundamental biochemical cause of galactosemia is often failure of the affected protein to fold correctly and/or its instability. This has been demonstrated in all four types of the disease. Some variant forms of GALT have been shown to be more sensitive to protease digestion and thermal denaturation than the wild type, thus supporting mutational effects on local and global stabilities of the enzymes [81,82,83]. Although only a minority of variants have been tested, computational studies suggest conformational instability may be a general effect for the majority of disease associated variants [84,85]. However, it should be noted that some disease associated variants (e.g., p.D28Y, p.L74P and p.F171S) are more stable against proteolysis and thermal denaturation, suggesting that some degree of flexibility is required for efficient catalysis and regulation [81]. Variants of galactokinase which are associated with the most severe forms are insoluble on expression in E. coli [86,87]. This suggests that they failed to fold and aggregated. The prokaryotic cellular environment is different to its eukaryotic equivalents, and so some caution is required in generalising this conclusion to human cells. Nevertheless, it is likely these variants fold less efficiently than the wild type. Some variants, including the one associated with the mildest known form of type II galactosemia (p.A198V), are expressed in reduced amounts by cells in comparison to the wild type [40,88]. Computational studies support the hypothesis that protein instability is a key factor in causing type II galactosemia [89,90]. Disease associated variants of UDP-galactose 4′ epimerase are typically less stable then the wild type and, in some cases, have reduced affinity for the NAD+ cofactor or aggregate in vivo [91,92,93,94,95,96]. Experimental and computational studies suggest there is a correlation between the degree of misfolding and the severity of the associated disease [92,97,98]. Similarly, in type IV galactosemia, experimental and computational studies link protein instability to reduced enzymatic activity and disease [30,31].
Taken together, these in silico, in vitro and in vivo data strongly suggest that misfolding is the main molecular mechanism in the pathology of galactosemia. Therefore, PCs may be an attractive therapy since they would help correct misfolding. However, the relatively mild symptoms normally associated with types II and IV, along with some forms of type III, may make it unrealistic to presume a PC strategy in these cases. The rarity of severe cases of type III may make the development of chaperones for this condition commercially unviable. Thus, type I represents the most likely form of the disease for which commercially and clinically sound PC strategy may be credible. Even in this type, the diversity of mutations may mean that PC treatments are only possible for a subset of variants. In particular, those variants with increased rigidity would be poor prospects for correction by chaperones. While protein misfolding is the most common fundamental cause of this disease, there are others, for example, mutations which directly affect the active site residues and prevent catalysis. Such variants would also not be addressed by a PC. The most common variants, p.S135L, Q188R, and K285N, in GALT are destabilised, and therefore represent viable targets for treatment [82,83]. Ideally, one molecule would be identified which corrects the folding of as many different disease-causing variants as possible. In Q188R, the most common variant in Caucasians, the mutation not only distorts the overall protein but severely distorts the active site [82,99]. The PC would need to correct this.
Initial studies suggested that non-specific binding of the amino acid arginine stabilises GALT [100]. Unfortunately, further work demonstrated that this is not the case [101]. In contrast, PCs bind to specific sites. This requires the identification of sites which contain sufficient exposed functional groups to allow respectable binding affinity and specificity. At least two such sites are potentially useful in human GALT. First, there are the two active sites. While it may seem counter intuitive to develop specific active site binding molecules since they are almost certain to act as competitive inhibitors, there are established cases of PC-like-molecules which bind to active sites (e.g., [102]). In the case of GALT, it was established experimentally that binding to the substrates increases the thermal stability of the protein [81]. Given that GALT has two active sites, if one site is occupied by a stabilising molecule (with affinity in the range of the natural substrate/coenzyme), this would stabilise the overall assembly and increase the catalytic efficiency of the second active site. The second potential chaperone binding site was identified computationally in a model of the enzyme, and its existence was confirmed in the crystal structure [81,82]. This pocket may act as a binding site for allosteric modifiers of the enzyme, but no such modifiers have been identified. GALT demonstrates cooperativity towards its substrates under some conditions in vitro, suggesting the possibility of allosteric communication within the enzyme [81].
4. Towards the Discovery of Pharmacological Chaperones for Galactosemia
The procedures to identify lead compounds as potential PCs can be widely classified in two groups: experimental and computational-structure guided screenings [103,104,105,106]. The possible route to finding PCs in galactosemia is illustrated in Figure 3.
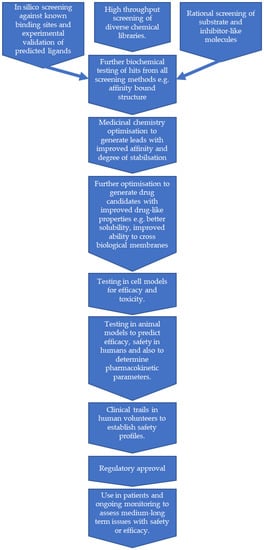
Figure 3.
A flow chart showing a possible route for pharmacological chaperones for galactosemia. This represents an idealised view, in reality some steps may need to be repeated and others performed in parallel.
4.1. Experimental Approaches
Experimental procedures require a setup for high-throughput screening that uses a suitable probe for the interaction between the target protein and the ligand. In this group, we can find different approaches depending on the physical and biological properties of the target protein (e.g., soluble vs. membrane protein, folded vs. natively unfolded protein, protein with enzymatic activity and/or allosteric regulation).
A general and classical approach for soluble proteins, either folded or natively unfolded proteins, is the thermal up-shift assay (more recently, this approach has been developed for membrane proteins) [105,107]. The rationale for this approach is straightforward: preferential binding of a ligand to the native state of the protein (i.e., with higher affinity for the native state) will increase its stability in comparison to those of non-native states. Note that this premise universally holds for equilibrium unfolding. However, it can also be extended in many cases for irreversibly denaturing proteins, as long as ligand binding provides significant kinetic stabilization upon binding to the folded state [108,109,110]. Therefore, it is relatively straightforward to develop high-throughput screening assays in which hits are identified by the increasing thermal stability of the protein, allowing for experimental uncertainty. This approach has been successfully applied to many protein targets in recent years [111,112,113]. Since the hits result in the stabilization of the protein in vitro, we can normally expect that this stabilization will translate into stabilization inside the cell, preventing protein aggregation or degradation of misfolded variants. This is reasonable because the destabilizing effect of mutations required for triggering protein degradation is rather low (about 8–12 kJ·mol−1), well within the achievable ligand-induced stabilization [114,115,116,117,118,119]. In other cases, protein degradation may depend on local destabilization of the protein structure, which initiates degradation, rather than in global destabilization [120,121,122,123]. Consequently, a disease-associated variant may bind and become globally (i.e. thermally) stabilized upon ligand binding, but this binding event may not affect the initiation site for degradation and thus will not be able to rescue the activity [123,124]. The relationships between global and local stability dynamics resulting in loss-of-function mechanisms in galactosemias are, to the best of our knowledge, not well-known. However, enhanced sampling of non-native, dynamic and partially unfolded conformations has been described for GALE disease-associated variants. These lead to reduced catalytic performance, and these fluctuations are modulated by native-state ligands such as NAD+ [125,126]. We may anticipate that the enhanced fluctuations in the native state due to disease-associated variants could also hold for degradation-prone variants. Thus, the identification of novel lead compounds (particularly those not binding to active site, or allosteric ligands) could be of pharmacological use to treat GALE deficiency [126] as well as other galactosemias.
A second approach to identify hits for the treatment of galactosemias is enzymatic assays. Like the thermal up-shift assays, these can be readily setup for high-throughput screening. Either ligands that inhibit the activity (but stabilize the enzyme) or those that restore the activity of variants by acting as allosteric chaperones could be identified.
A third group of assays are those based on screening in eukaryotic cells [106]. These assays are potentially more problematic since handling eukaryotic cells is usually more challenging than working with purified proteins. However, several good models of galactosemia-causing variants have been developed in budding yeast (S. cerevisiae). These allow the straightforward measurement of enzyme activity and protein levels, which are related to protein stability [81,91,94]. Thus, these models represent an excellent starting point to develop high-throughput screens [127,128].
An alternative method to high throughput screening is the fragment-based approach [129,130]. This requires protein crystals which are soaked with small organic molecules and the resulting structure solved. The organic molecules are chosen for their similarity to common building blocks (e.g., acetane, phenol) present in more complex molecules. Although these small molecules typically bind with low affinity, superimposition of the structures with different small molecules bound enables the prediction of more complex molecules. These are more likely to bind with high affinity and specificity. Further improvements in both of these attributes can be achieved through conventional medicinal chemistry.
4.2. Computational Approaches
Computational approaches can be used to help identify possible binding sites and small molecules which may bind to these. This is achieved by using the protein’s three-dimensional structure to screen potential small molecules. In theory, this process should be similar to screening for inhibitors or antagonists, which is routinely undertaken in drug discovery, but there are two main differences. First, in conventional drug discovery, the binding site is often already known (e.g., the active site of an enzyme or the ligand binding site of a receptor). Second, in the discovery of PCs, we aim to improve or restore function, whereas conventional drug discovery typically seeks to inhibit or block function. To address the first problem, there are programs which can identify possible binding pockets on the surface of proteins (see above for an example of this applied to GALT). Once a potential pocket has been identified, molecules can be screened using conventional methods. This will result in a prediction of molecules which will bind at the site ranked by estimated affinity. While many protein–ligand interactions result in the stabilization of the protein, it is not necessarily the highest affinity binders which will result in the greatest stabilization. Some insight into the degree of stabilization might be obtained from molecular dynamics simulations, but these are time consuming and it may be preferable to move directly to experimental measurements of stabilization. To the best of our knowledge, no computational screening for potential PCs has been carried out with any of the enzymes associated with galactosemia.
5. Potential Issues with Clinical Use
Ideally, the PC for type 1 galactosemia would be administered as soon as possible after birth. In the absence of pre-birth diagnosis, this is likely to be after the baby’s first milk feed. Rapid testing would then be required to establish a diagnosis, followed by sequencing of the GALT gene. If the drug had few side effects, then it might be given as soon as galactosemia was suspected. It could be discontinued in the event of a different diagnosis or if gene sequencing revealed the patient had a mutation which was unsuitable for the drug. In reality, the situation is likely to be more complicated, and there are some potential issues with the long-term use of PCs in this disease.
5.1. Screening, Testing and Sequencing
Pre-natal screening is routinely performed for a number of genetic conditions (e.g., cystic fibrosis) if a strong family history is present [84]. This would be ideal to allow the most rapid treatment, but this type of testing can be invasive (e.g., amniocentesis). It also presents ethical issues. This topic also raises the question as to when the phenotypic features of the disease first develop. While it is assumed that the majority of the disease processes occur postnatally, this is not known. Galactose, galactose 1-phosphate and galactitol have all been detected in the liver and amniotic fluid of foetuses at 20 weeks gestation. In some rare cases, newborns have been found to have cataracts, which would suggest some pathology can occur in utero [131,132,133]. Some aberrant glycosylation may occur in the foetus. This may be particularly harmful if it occurs in the brain or nervous system. Clearly, if PC strategy relies on being able to protect a healthy newborn, the benefits may be reduced if significant damage has been done in utero. If a prenatal diagnosis was made, there is the potential that in utero treatment with a PC could be commenced.
Newborn screening is performed in many countries for galactosemia. The timing of these tests varies between 24 h to several weeks post birth. In the most severe cases, the baby would have presented with severe symptoms before results are available. This type of screening may be most useful in detecting the milder phenotypes [36]. Delays occur in the testing and sequencing of potential galactosemia sufferers due to lack of capacity in many healthcare systems. Often, samples need to be shipped to tertiary sites for testing and results must be analysed before being sent back. All of this takes time and would delay treatment [85].
With over 300 mutations known to cause type 1 galactosemia, it is unlikely that the molecular mechanism resulting in disease will be identical in all cases. Experimental and computational evidence suggests that the majority of point mutations result in protein misfolding and, therefore, may be amenable to correction by PCs (see Section 3). However, mutations which affect active site residues, cause deletions, or cause defects in mRNA splicing will not be corrected by a PC. Some point mutations may also result in proteins which are refractory to having their structures restored by PCs, e.g., mutations which affect residues in the drug binding site result in increased rigidification of the protein or result in a protein which is so destabilised that the chaperone is insufficient to restore function. Sequencing will be vital to determine if the patient has at least one mutation which is amenable to the action of PCs.
5.2. Dosing
With most drugs, adult dosages are straightforward. However, as this drug would be used soon after birth, achieving a therapeutic and non-toxic dose will be difficult to ascertain, but this is important to its effectiveness. The drug dosage will need to be tailored to weight, age and/or body surface area and adjusted as the patient grows. It may also be necessary to tailor the dose to the individual patient’s response as metabolism will vary between individuals, as people mature at different rates. Thus, doses may need to be personalised and are likely to vary until the patient reaches adulthood or beyond. There is some evidence that the dietary restriction of galactose can be relaxed in adult patients [86]. Therefore, a reduced dose may be effective in some adult patients. Ideally, the effectiveness of the dose would be monitored and adjusted according to blood galactose 1-phosphate levels. Other biomarkers such as the incorporation of galactose into glycoproteins may also be useful [87]. Given the risk of toxicity (see below), it may be necessary to check for effects on liver and kidney function (serum urea and electrolytes and liver function tests).
Initially, the only source of dosing information will be from clinical trials, which are likely to have relatively small numbers of patients enrolled due to the rarity of the disease. As with all potential drugs for use in children, clinical trials are very challenging due to ethical approvals and challenges with recruitment. It would be difficult to extrapolate doses for all children at different maturities based on very limited trial data. As a consequence, it will be important that post trial data on dosing in initial recipients are shared effectively. The existence of patient registries such as GalNet may help keep in contact with post trial patients for longer [88].
5.3. Toxicity
All drugs have side effects. These can arise through interaction with non-target biomolecules, toxic metabolites or unwanted effects resulting from interaction with the target. Since the PC would increase GALT activity, it is unlikely there would be unwanted consequences of this action; the other two causes are possible.
Many side effects are relatively mild in most people or can be mitigated by other drugs. However, drugs which cause lasting harm due to toxicity can normally only be used for limited periods and are rarely used in children. There are potential effects on development or lifelong harm. A PC for galactosemia is likely to be started in the first weeks of life and continued throughout the life of the patient. Therefore, cumulative toxicity could be an issue as lipophilic drugs will build up in adipose tissue over time. Since clinical trials are carried out over a limited period, not all toxicities will be apparent at the time of publishing and will only become apparent after years of follow up.
There is a lack of data on the galactosemic phenotype in older patients; most studies focus on children or adults under 30 years old. Therefore, it may be difficult to determine whether symptoms are due to disease phenotype or potential drug toxicities. In this population, there is also a greater chance of comorbidities and interactions with the drugs used to treat these.
5.4. Costs and Returns on Investments
Drug discovery is expensive and is normally financed by the profits derived from the sale of existing drugs. This means that pharmaceutical companies may be reluctant to invest in drugs to treat rare diseases such as galactosemia. This problem can be partly alleviated by governments or charities sharing costs and risks. Any PC for galactosemia is likely to be granted orphan drug status. The definition of orphan drug differs depending on the jurisdiction, but it is largely dependent on the disease being rare and therefore a drug having little profitability, but which would meet a public health need. Orphan drug status can have benefits such as tax incentives, direct government subsidiary, increased patent time or a streamlined approval process [89].
Nevertheless, return on investment for pharmaceutical companies is likely to occur over the longer term (50+ years), if at all. This will require long-term strategies by the pharmaceutical industry which are likely to extend beyond the complete turnover of their boards of directors. Initially, the likely lack of competition may encourage companies to develop these kinds of drugs. However, this lack of competition may mean other companies do not seek to develop rival drugs with improved efficacy.
6. Conclusions
As in many other rare, inherited metabolic diseases, galactosemias lack adequate treatments or cures. Future research must focus on identifying safe and efficient new treatment options for these patients. Despite the significant genetic diversity found in all types of galactosemias, most mutations impact the stability of the corresponding protein or its ability to fold properly in vivo. Consequently, the development of PCs has emerged as a plausible approach for future therapies in galactosemias. Easy recombinant production and in vitro characterization of wild type and mutant variant proteins, the existence of simple and easy to handle eukaryotic expression systems (e.g., budding yeast), and the availability of high resolution crystal structures will hopefully allow the identification of potential PCs for these diseases, using different types of in vitro and in silico approaches.
An effective PC for galactosemia would be a significant improvement to the current treatment mantra of maintaining a galactose-free diet. If started soon after birth, using a PC to restore some enzyme function could be life-changing for patients, improving both their morbidity and mortality by helping to avoid harmful complications such as liver dysfunction and cataracts.
Author Contributions
S.B.: drafted Section 1, Section 5, Section 6 and Figure 1, Figure 2 and Figure 3; T.J.M.: drafted Section 4.1; A.L.P.: drafted Section 4.1 and Section 6; D.J.T.: drafted Section 3, Section 4.1, Section 4.2 and Section 5; all authors edited and approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the ERDF/Spanish Ministry of Science, Innovation and Universities—State Research Agency (Grant RTI2018-096246-B-I00, to A.L.P.) and FEDER/Junta de Andalucía-Consejería de Transformación Económica, Industria, Conocimiento y Universidades (Grant P18-RT-2413, to A.L.P.). The funding sources had no role in the design of this study, analysis of data or preparation of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Timson, D.J. The molecular basis of galactosemia—Past, present and future. Gene 2016, 589, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J. Molecular Genetics of Galactosaemia. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2017. [Google Scholar] [CrossRef]
- Fridovich-Keil, J.L. Galactosemia: The good, the bad, and the unknown. J. Cell. Physiol. 2006, 209, 701–705. [Google Scholar] [CrossRef]
- Fridovich-Keil, J.L.; Walter, J.H. Galactosemia. In The Online Metabolic and Molecular Bases of Inherited Diseases; Valle, D., Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Eds.; McGraw-Hill: New York, NY, USA, 2008. [Google Scholar]
- Demirbas, D.; Coelho, A.I.; Rubio-Gozalbo, M.E.; Berry, G.T. Hereditary galactosemia. Metabolism 2018, 83, 188–196. [Google Scholar] [CrossRef]
- Maratha, A.; Stockmann, H.; Coss, K.P.; Estela Rubio-Gozalbo, M.; Knerr, I.; Fitzgibbon, M.; McVeigh, T.P.; Foley, P.; Moss, C.; Colhoun, H.O.; et al. Classical galactosaemia: Novel insights in IgG N-glycosylation and N-glycan biosynthesis. Eur. J. Hum. Genet. 2016, 24, 976–984. [Google Scholar] [CrossRef]
- Holden, H.M.; Rayment, I.; Thoden, J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem. 2003, 278, 43885–43888. [Google Scholar] [CrossRef]
- Frey, P.A. The Leloir pathway: A mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 461–470. [Google Scholar] [CrossRef]
- Caputto, R.; Leloir, L.F.; Trucco, R.E.; Cardini, C.E.; Paladini, A.C. The enzymatic transformation of galactose into glucose derivatives. J. Biol. Chem. 1949, 179, 497–498. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Potter, N.L.; Gordon, C.M.; Green, R.C.; Greenstein, P.; Gubbels, C.S.; Rubio-Gozalbo, E.; Schomer, D.; Welt, C.; Anastasoaie, V.; et al. The adult galactosemic phenotype. J. Inherit. Metab. Dis. 2012, 35, 279–286. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Haskovic, M.; Bosch, A.M.; Burnyte, B.; Coelho, A.I.; Cassiman, D.; Couce, M.L.; Dawson, C.; Demirbas, D.; Derks, T.; et al. The natural history of classic galactosemia: Lessons from the GalNet registry. Orphanet J. Rare Dis. 2019, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, A.; Grunewald, S.; Murphy, E.; Coenen, M.A.; Eggink, H.; Zutt, R.; Rubio-Gozalbo, M.E.; Bosch, A.M.; Williams, M.; Derks, T.G.J.; et al. Movement disorders and nonmotor neuropsychological symptoms in children and adults with classical galactosemia. J. Inherit. Metab. Dis. 2019, 42, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, C.S.; Land, J.A.; Evers, J.L.; Bierau, J.; Menheere, P.P.; Robben, S.G.; Rubio-Gozalbo, M.E. Primary ovarian insufficiency in classic galactosemia: Role of FSH dysfunction and timing of the lesion. J. Inherit. Metab. Dis. 2013, 36, 29–34. [Google Scholar] [CrossRef]
- Fridovich-Keil, J.L.; Gubbels, C.S.; Spencer, J.B.; Sanders, R.D.; Land, J.A.; Rubio-Gozalbo, E. Ovarian function in girls and women with GALT-deficiency galactosemia. J. Inherit. Metab. Dis. 2011, 34, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Welling, L.; Bernstein, L.E.; Berry, G.T.; Burlina, A.B.; Eyskens, F.; Gautschi, M.; Grunewald, S.; Gubbels, C.S.; Knerr, I.; Labrune, P.; et al. International clinical guideline for the management of classical galactosemia: Diagnosis, treatment, and follow-up. J. Inherit. Metab. Dis. 2017, 40, 171–176. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Timson, D.J. Structural and molecular biology of type I galactosemia: Disease-associated mutations. IUBMB Life 2011, 63, 949–954. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Timson, D.J. The structural and molecular biology of type I galactosemia: Enzymology of galactose 1-phosphate uridylyltransferase. IUBMB Life 2011, 63, 694–700. [Google Scholar] [CrossRef]
- Flanagan, J.M.; McMahon, G.; Brendan Chia, S.H.; Fitzpatrick, P.; Tighe, O.; O’Neill, C.; Briones, P.; Gort, L.; Kozak, L.; Magee, A.; et al. The role of human demographic history in determining the distribution and frequency of transferase-deficient galactosaemia mutations. Heredity 2010, 104, 148–154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tyfield, L.; Reichardt, J.; Fridovich-Keil, J.; Croke, D.T.; Elsas, L.J., II; Strobl, W.; Kozak, L.; Coskun, T.; Novelli, G.; Okano, Y.; et al. Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene. Hum. Mutat. 1999, 13, 417–430. [Google Scholar] [CrossRef]
- Wells, L.; Fridovich-Keil, J.L. Biochemical characterization of the S135L allele of galactose-1-phosphate uridylyltransferase associated with galactosaemia. J. Inherit. Metab. Dis. 1997, 20, 633–642. [Google Scholar] [CrossRef]
- Lai, K.; Elsas, L.J. Structure-function analyses of a common mutation in blacks with transferase-deficiency galactosemia. Mol. Genet. Metab. 2001, 74, 264–272. [Google Scholar] [CrossRef]
- Lai, K.; Langley, S.D.; Singh, R.H.; Dembure, P.P.; Hjelm, L.N.; Elsas, L.J., II. A prevalent mutation for galactosemia among black Americans. J. Pediatr. 1996, 128, 89–95. [Google Scholar] [CrossRef]
- Holden, H.M.; Thoden, J.B.; Timson, D.J.; Reece, R.J. Galactokinase: Structure, function and role in type II galactosemia. Cell. Mol. Life Sci. CMLS 2004, 61, 2471–2484. [Google Scholar] [CrossRef]
- Timson, D.J.; Reece, R.J.; Thoden, J.B.; Holden, H.M. Galactokinase Deficiency. In Encyclopedia of Molecular Mechanisms of Disease; Lang, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 679–680. [Google Scholar]
- Bosch, A.M.; Bakker, H.D.; van Gennip, A.H.; van Kempen, J.V.; Wanders, R.J.; Wijburg, F.A. Clinical features of galactokinase deficiency: A review of the literature. J. Inherit. Metab. Dis. 2002, 25, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J. The structural and molecular biology of type III galactosemia. IUBMB Life 2006, 58, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Fridovich-Keil, J.; Bean, L.; He, M.; Schroer, R. Epimerase Deficiency Galactosemia. In GeneReviews; Pagon, R.A., Bird, T.D., Dolan, C.R., Stephens, K., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Openo, K.K.; Schulz, J.M.; Vargas, C.A.; Orton, C.S.; Epstein, M.P.; Schnur, R.E.; Scaglia, F.; Berry, G.T.; Gottesman, G.S.; Ficicioglu, C.; et al. Epimerase-deficiency galactosemia is not a binary condition. Am. J. Hum. Genet. 2006, 78, 89–102. [Google Scholar] [CrossRef]
- Daenzer, J.M.; Sanders, R.D.; Hang, D.; Fridovich-Keil, J.L. UDP-galactose 4′-epimerase activities toward UDP-Gal and UDP-GalNAc play different roles in the development of Drosophila melanogaster. PLoS Genet. 2012, 8, e1002721. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, S.; Kikuchi, A.; Wada, Y.; Arai-Ichinoi, N.; Sakamoto, O.; Tamiya, G.; Kure, S. The prevalence of GALM mutations that cause galactosemia: A database of functionally evaluated variants. Mol. Genet. Metab. 2019, 126, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Kikuchi, A.; Arai-Ichinoi, N.; Sakamoto, O.; Takezawa, Y.; Iwasawa, S.; Niihori, T.; Nyuzuki, H.; Nakajima, Y.; Ogawa, E.; et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet. Med. 2019, 21, 1286–1294. [Google Scholar] [CrossRef]
- Timson, D.J. Type IV galactosemia. Genet. Med. 2019, 21, 1283–1285. [Google Scholar] [CrossRef]
- Banford, S.; Timson, D.J. The structural and molecular biology of type IV galactosemia. Biochimie 2020. [Google Scholar] [CrossRef]
- Beutler, E.; Baluda, M.C. A simple spot screening test for galactosemia. J. Lab. Clin. Med. 1966, 68, 137–141. [Google Scholar]
- Ohlsson, A.; Guthenberg, C.; von Döbeln, U. Galactosemia screening with low false-positive recall rate: The Swedish experience. JIMD Rep. 2012, 2, 113–117. [Google Scholar] [CrossRef]
- Gitzelmann, R. Estimation of galactose-I-phosphate in erythrocytes: A rapid and simple enzymatic method. Clin. Chim. Acta 1969, 26, 313–316. [Google Scholar] [CrossRef]
- Cohen, A.S.; Baurek, M.; Lund, A.M.; Dunø, M.; Hougaard, D.M. Including Classical Galactosaemia in the Expanded Newborn Screening Panel Using Tandem Mass Spectrometry for Galactose-1-Phosphate. Int. J. Neonatal Screen. 2019, 5, 19. [Google Scholar] [CrossRef]
- Li, Y.; Ptolemy, A.S.; Harmonay, L.; Kellogg, M.; Berry, G.T. Quantification of galactose-1-phosphate uridyltransferase enzyme activity by liquid chromatography-tandem mass spectrometry. Clin. Chem. 2010, 56, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kotb, M.A.; Mansour, L.; Shamma, R.A. Screening for galactosemia: Is there a place for it? Int. J. Gen. Med. 2019, 12, 193–205. [Google Scholar] [CrossRef]
- Okano, Y.; Asada, M.; Fujimoto, A.; Ohtake, A.; Murayama, K.; Hsiao, K.J.; Choeh, K.; Yang, Y.; Cao, Q.; Reichardt, J.K.; et al. A genetic factor for age-related cataract: Identification and characterization of a novel galactokinase variant, “Osaka” in Asians. Am. J. Hum. Genet. 2001, 68, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.T.; Holton, J.B.; Lennox, A.C.; Hodges, I.C. Early morning urine galactitol levels in relation to galactose intake: A possible method of monitoring the diet in galactokinase deficiency. J. Inherit. Metab. Dis. 1988, 11 (Suppl. S2), 243–245. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Fridovich-Keil, J.L. The yeast, Saccharomyces cerevisiae, as a model system for the study of human genetic disease. SAAS Bull. Biochem. Biotechnol. 1996, 9, 83–88. [Google Scholar]
- Riehman, K.; Crews, C.; Fridovich-Keil, J.L. Relationship between genotype, activity, and galactose sensitivity in yeast expressing patient alleles of human galactose-1-phosphate uridylyltransferase. J. Biol. Chem. 2001, 276, 10634–10640. [Google Scholar] [CrossRef]
- Ross, K.L.; Davis, C.N.; Fridovich-Keil, J.L. Differential roles of the Leloir pathway enzymes and metabolites in defining galactose sensitivity in yeast. Mol. Genet. Metab. 2004, 83, 103–116. [Google Scholar] [CrossRef]
- Wasilenko, J.; Fridovich-Keil, J.L. Relationship between UDP-galactose 4′-epimerase activity and galactose sensitivity in yeast. J. Biol. Chem. 2006, 281, 8443–8449. [Google Scholar] [CrossRef] [PubMed]
- Mumma, J.O.; Chhay, J.S.; Ross, K.L.; Eaton, J.S.; Newell-Litwa, K.A.; Fridovich-Keil, J.L. Distinct roles of galactose-1P in galactose-mediated growth arrest of yeast deficient in galactose-1P uridylyltransferase (GALT) and UDP-galactose 4′-epimerase (GALE). Mol. Genet. Metab. 2008, 93, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Kushner, R.F.; Ryan, E.L.; Sefton, J.M.; Sanders, R.D.; Lucioni, P.J.; Moberg, K.H.; Fridovich-Keil, J.L. A Drosophila melanogaster model of classic galactosemia. Dis. Model. Mech. 2010, 3, 618–627. [Google Scholar] [CrossRef]
- Sanders, R.D.; Sefton, J.M.; Moberg, K.H.; Fridovich-Keil, J.L. UDP-galactose 4′ epimerase (GALE) is essential for development of Drosophila melanogaster. Dis. Model. Mech. 2010, 3, 628–638. [Google Scholar] [CrossRef]
- Daenzer, J.M.; Fridovich-Keil, J.L. Drosophila melanogaster Models of Galactosemia. Curr. Top. Dev. Biol. 2017, 121, 377–395. [Google Scholar] [CrossRef]
- Tang, M.; Siddiqi, A.; Witt, B.; Yuzyuk, T.; Johnson, B.; Fraser, N.; Chen, W.; Rascon, R.; Yin, X.; Goli, H.; et al. Subfertility and growth restriction in a new galactose-1 phosphate uridylyltransferase (GALT)-deficient mouse model. Eur. J. Hum. Genet. EJHG 2014, 22, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Brokate-Llanos, A.M.; Monje, J.M.; Murdoch Pdel, S.; Muñoz, M.J. Developmental defects in a Caenorhabditis elegans model for type III galactosemia. Genetics 2014, 198, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Vanoevelen, J.M.; van Erven, B.; Bierau, J.; Huang, X.; Berry, G.T.; Vos, R.; Coelho, A.I.; Rubio-Gozalbo, M.E. Impaired fertility and motor function in a zebrafish model for classic galactosemia. J. Inherit. Metab. Dis. 2018, 41, 117–127. [Google Scholar] [CrossRef]
- Haskovic, M.; Coelho, A.I.; Lindhout, M.; Zijlstra, F.; Veizaj, R.; Vos, R.; Vanoevelen, J.M.; Bierau, J.; Lefeber, D.J.; Rubio-Gozalbo, M.E. Nucleotide sugar profiles throughout development in wildtype and galt knockout zebrafish. J. Inherit. Metab. Dis. 2020, 43, 994–1001. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Daenzer, J.M.I.; MacWilliams, J.A.; Head, S.T.; Williams, M.B.; Geurts, A.M.; Schroeder, J.P.; Weinshenker, D.; Fridovich-Keil, J.L. A galactose-1-phosphate uridylyltransferase-null rat model of classic galactosemia mimics relevant patient outcomes and reveals tissue-specific and longitudinal differences in galactose metabolism. J. Inherit. Metab. Dis. 2020, 43, 518–528. [Google Scholar] [CrossRef]
- Lai, K.; Elsas, L.J.; Wierenga, K.J. Galactose toxicity in animals. IUBMB Life 2009, 61, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Schadewaldt, P.; Kamalanathan, L.; Hammen, H.-W.; Kotzka, J.; Wendel, U. Endogenous galactose formation in galactose-1-phosphate uridyltransferase deficiency. Arch. Physiol. Biochem. 2014, 120, 228–239. [Google Scholar] [CrossRef]
- Berry, G.T.; Nissim, I.; Lin, Z.; Mazur, A.T.; Gibson, J.B.; Segal, S. Endogenous synthesis of galactose in normal men and patients with hereditary galactosaemia. Lancet 1995, 346, 1073–1074. [Google Scholar] [CrossRef]
- Berry, G.T.; Moate, P.J.; Reynolds, R.A.; Yager, C.T.; Ning, C.; Boston, R.C.; Segal, S. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol. Genet. Metab. 2004, 81, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J. Repurposing drugs for the treatment of galactosemia. Expert Opin. Orphan Drugs 2019, 7, 443–451. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Timson, D.J. Galactosemia: Opportunities for novel therapies. In Protein Homeostasis Diseases: Mechanisms and Novel Therapies; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Timson, D.J. Therapies for galactosemia: A patent landscape. Pharm. Pat. Anal. 2020, 9, 45–51. [Google Scholar] [CrossRef]
- Lai, K.; Boxer, M.B.; Marabotti, A. GALK inhibitors for classic galactosemia. Future Med. Chem. 2014, 6, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Delnoy, B.; Coelho, A.I.; Rubio-Gozalbo, M.E. Current and Future Treatments for Classic Galactosemia. J. Pers. Med. 2021, 11, 75. [Google Scholar] [CrossRef]
- Brandvold, K.R.; Morimoto, R.I. The Chemical Biology of Molecular Chaperones-Implications for Modulation of Proteostasis. J. Mol. Biol. 2015, 427, 2931–2947. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Muntau, A.C.; Leandro, J.; Staudigl, M.; Mayer, F.; Gersting, S.W. Innovative strategies to treat protein misfolding in inborn errors of metabolism: Pharmacological chaperones and proteostasis regulators. J. Inherit. Metab. Dis. 2014, 37, 505–523. [Google Scholar] [CrossRef]
- Ringe, D.; Petsko, G.A. What are pharmacological chaperones and why are they interesting? J. Biol. 2009, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.-X.; Conn, P.M. Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Leidenheimer, N.J.; Ryder, K.G. Pharmacological chaperoning: A primer on mechanism and pharmacology. Pharmacol. Res. 2014, 83, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.E.; Kelly, J.W. Therapeutic approaches to protein-misfolding diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Porto, C.; Cardone, M.; Fontana, F.; Rossi, B.; Tuzzi, M.R.; Tarallo, A.; Barone, M.V.; Andria, G.; Parenti, G. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol. Ther. 2009, 17, 964–971. [Google Scholar] [CrossRef]
- Fan, J.-Q. A counterintuitive approach to treat enzyme deficiencies: Use of enzyme inhibitors for restoring mutant enzyme activity. Biol. Chem. 2008, 389, 1–11. [Google Scholar] [CrossRef]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef]
- Orwig, S.D.; Tan, Y.L.; Grimster, N.P.; Yu, Z.; Powers, E.T.; Kelly, J.W.; Lieberman, R.L. Binding of 3,4,5,6-tetrahydroxyazepanes to the acid-β-glucosidase active site: Implications for pharmacological chaperone design for Gaucher disease. Biochemistry 2011, 50, 10647–10657. [Google Scholar] [CrossRef][Green Version]
- Fraser-Pitt, D.; O’Neil, D. Cystic fibrosis—A multiorgan protein misfolding disease. Future Sci. OA 2015, 1, FSO57. [Google Scholar] [CrossRef]
- Rowe, S.M.; Verkman, A.S. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb. Perspect. Med. 2013, 3, a009761. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Bulawa, C.E.; Connelly, S.; Devit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Gleason, T.J.; Fridovich-Keil, J.L.; Timson, D.J. Misfolding of galactose 1-phosphate uridylyltransferase can result in type I galactosemia. Biochim. Biophys. Acta 2013, 1832, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Kopec, J.; Pey, A.L.; Fitzpatrick, F.; Patel, D.; Chalk, R.; Shrestha, L.; Yue, W.W. Molecular basis of classic galactosemia from the structure of human galactose 1-phosphate uridylyltransferase. Hum. Mol. Genet. 2016, 25, 2234–2244. [Google Scholar] [CrossRef]
- Coelho, A.I.; Trabuco, M.; Ramos, R.; Silva, M.J.; Tavares de Almeida, I.; Leandro, P.; Rivera, I.; Vicente, J.B. Functional and structural impact of the most prevalent missense mutations in classic galactosemia. Mol. Genet. Genom. Med. 2014, 2, 484–496. [Google Scholar] [CrossRef]
- D’acierno, A.; Facchiano, A.; Marabotti, A. GALT protein database, a bioinformatics resource for the management and analysis of structural features of a galactosemia-related protein and its mutants. Genom. Proteom. Bioinform. Beijing Genom. Inst. 2009, 7, 71–76. [Google Scholar] [CrossRef]
- D’Acierno, A.; Facchiano, A.; Marabotti, A. GALT protein database: Querying structural and functional features of GALT enzyme. Hum. Mutat. 2014, 35, 1060–1067. [Google Scholar] [CrossRef]
- Timson, D.J.; Reece, R.J. Functional analysis of disease-causing mutations in human galactokinase. Eur. J. Biochem. FEBS 2003, 270, 1767–1774. [Google Scholar] [CrossRef]
- Sangiuolo, F.; Magnani, M.; Stambolian, D.; Novelli, G. Biochemical characterization of two GALK1 mutations in patients with galactokinase deficiency. Hum. Mutat. 2004, 23, 396. [Google Scholar] [CrossRef] [PubMed]
- Park, H.D.; Bang, Y.L.; Park, K.U.; Kim, J.Q.; Jeong, B.H.; Kim, Y.S.; Song, Y.H.; Song, J. Molecular and biochemical characterization of the GALK1 gene in Korean patients with galactokinase deficiency. Mol. Genet. Metab 2007, 91, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Sneha, P.; Ebrahimi, E.A.; Ghazala, S.A.; Thirumal Kumar, D.; Siva, R.; George, P.D.C.; Zayed, H. Structural analysis of missense mutations in galactokinase 1 (GALK1) leading to galactosemia type-2. J. Cell. Biochem. 2018, 119, 7585–7598. [Google Scholar] [CrossRef]
- Jojart, B.; Szori, M.; Izsak, R.; Marsi, I.; Laszlo, A.; Csizmadia, I.G.; Viskolcz, B. The effect of a Pro(28)Thr point mutation on the local structure and stability of human galactokinase enzyme-a theoretical study. J. Mol. Model. 2011, 17, 2639–2649. [Google Scholar] [CrossRef]
- Chhay, J.S.; Vargas, C.A.; McCorvie, T.J.; Fridovich-Keil, J.L.; Timson, D.J. Analysis of UDP-galactose 4′-epimerase mutations associated with the intermediate form of type III galactosemia. J. Inherit. Metab. Dis. 2008, 31, 108–116. [Google Scholar] [CrossRef]
- Timson, D.J. Functional analysis of disease-causing mutations in human UDP-galactose 4-epimerase. FEBS J. 2005, 272, 6170–6177. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Wasilenko, J.; Liu, Y.; Fridovich-Keil, J.L.; Timson, D.J. In vivo and in vitro function of human UDP-galactose 4′-epimerase variants. Biochimie 2011, 93, 1747–1754. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Liu, Y.; Frazer, A.; Gleason, T.J.; Fridovich-Keil, J.L.; Timson, D.J. Altered cofactor binding affects stability and activity of human UDP-galactose 4′-epimerase: Implications for type III galactosemia. Biochim. Biophys. Acta 2012, 1822, 1516–1526. [Google Scholar] [CrossRef]
- Paul, S.; McCorvie, T.J.; Zschocke, J.; Timson, D.J. Disturbed cofactor binding by a novel mutation in UDP-galactose 4′-epimerase results in a type III galactosemia phenotype at birth. RSC Adv. 2016, 6, 17297–17301. [Google Scholar] [CrossRef]
- Bang, Y.L.; Nguyen, T.T.; Trinh, T.T.; Kim, Y.J.; Song, J.; Song, Y.H. Functional analysis of mutations in UDP-galactose-4-epimerase (GALE) associated with galactosemia in Korean patients using mammalian GALE-null cells. FEBS J. 2009, 276, 1952–1961. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Timson, D.J. In silico prediction of the effects of mutations in the human UDP-galactose 4′-epimerase gene: Towards a predictive framework for type III galactosemia. Gene 2013, 524, 95–104. [Google Scholar] [CrossRef]
- Timson, D.J.; Lindert, S. Comparison of dynamics of wildtype and V94M human UDP-galactose 4-epimerase-A computational perspective on severe epimerase-deficiency galactosemia. Gene 2013, 526, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Marabotti, A.; Facchiano, A.M. Homology modeling studies on human galactose-1-phosphate uridylyltransferase and on its galactosemia-related mutant Q188R provide an explanation of molecular effects of the mutation on homo- and heterodimers. J. Med. Chem. 2005, 48, 773–779. [Google Scholar] [CrossRef]
- Coelho, A.I.; Trabuco, M.; Silva, M.J.; de Almeida, I.T.; Leandro, P.; Rivera, I.; Vicente, J.B. Arginine Functionally Improves Clinically Relevant Human Galactose-1-Phosphate Uridylyltransferase (GALT) Variants Expressed in a Prokaryotic Model. JIMD Rep. 2015, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Haskovic, M.; Derks, B.; van der Ploeg, L.; Trommelen, J.; Nyakayiru, J.; van Loon, L.J.C.; Mackinnon, S.; Yue, W.W.; Peake, R.W.A.; Zha, L.; et al. Arginine does not rescue p.Q188R mutation deleterious effect in classic galactosemia. Orphanet J. Rare Dis. 2018, 13, 212. [Google Scholar] [CrossRef]
- Strandback, E.; Lienhart, W.D.; Hromic-Jahjefendic, A.; Bourgeois, B.; Högler, A.; Waltenstorfer, D.; Winkler, A.; Zangger, K.; Madl, T.; Gruber, K.; et al. A small molecule chaperone rescues the stability and activity of a cancer-associated variant of NAD(P)H:quinone oxidoreductase 1 in vitro. FEBS Lett. 2020, 594, 424–438. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Yue, W.W. Structure-guided discovery of pharmacological chaperones targeting protein conformational and misfolding diseases. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 281–308. [Google Scholar]
- Rizzuti, B.; Grande, F. Virtual screening in drug discovery: A precious tool for a still-demanding challenge. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 309–328. [Google Scholar]
- Støve, S.I.; Flydal, M.I.; Hausvik, E.; Underhaug, J.; Martinez, A. Differential scanning fluorimetry in the screening and validation of pharmacological chaperones for soluble and membrane proteins. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 329–342. [Google Scholar]
- Janovick, J.A.; Ulloa-Aguirre, A. Cellular high-throughput screening. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 343–358. [Google Scholar]
- Abian, O.; Vega, S.; Neira, J.L.; Velazquez-Campoy, A. High-throughput screening for intrinsically disordered proteins by using biophysical methods. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 359–388. [Google Scholar]
- Pey, A.L. Protein homeostasis disorders of key enzymes of amino acids metabolism: Mutation-induced protein kinetic destabilization and new therapeutic strategies. Amino Acids 2013, 45, 1331–1341. [Google Scholar] [CrossRef]
- Pey, A.L.; Majtan, T.; Sanchez-Ruiz, J.M.; Kraus, J.P. Human cystathionine beta-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): Complex regulation of CBS activity and stability by SAM. Biochem. J. 2013, 449, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ruiz, J.M. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry. Biophys. J. 1992, 61, 921–935. [Google Scholar] [CrossRef]
- Santofimia-Castano, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velazquez-Campoy, A.; Abian, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef]
- Pey, A.L.; Ying, M.; Cremades, N.; Velazquez-Campoy, A.; Scherer, T.; Thony, B.; Sancho, J.; Martinez, A. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J. Clin. Investig. 2008, 118, 2858–2867. [Google Scholar] [CrossRef]
- Jorge-Finnigan, A.; Brasil, S.; Underhaug, J.; Ruiz-Sala, P.; Merinero, B.; Banerjee, R.; Desviat, L.R.; Ugarte, M.; Martinez, A.; Perez, B. Pharmacological chaperones as a potential therapeutic option in methylmalonic aciduria cblB type. Hum. Mol. Genet. 2013, 22, 3680–3689. [Google Scholar] [CrossRef]
- Pey, A.L.; Stricher, F.; Serrano, L.; Martinez, A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am. J. Hum. Genet. 2007, 81, 1006–1024. [Google Scholar] [CrossRef]
- Abildgaard, A.B.; Stein, A.; Nielsen, S.V.; Schultz-Knudsen, K.; Papaleo, E.; Shrikhande, A.; Hoffmann, E.R.; Bernstein, I.; Gerdes, A.M.; Takahashi, M.; et al. Computational and cellular studies reveal structural destabilization and degradation of MLH1 variants in Lynch syndrome. eLife 2019, 8. [Google Scholar] [CrossRef]
- Nielsen, S.V.; Stein, A.; Dinitzen, A.B.; Papaleo, E.; Tatham, M.H.; Poulsen, E.G.; Kassem, M.M.; Rasmussen, L.J.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Predicting the impact of Lynch syndrome-causing missense mutations from structural calculations. PLoS Genet. 2017, 13, e1006739. [Google Scholar] [CrossRef] [PubMed]
- Scheller, R.; Stein, A.; Nielsen, S.V.; Marin, F.I.; Gerdes, A.M.; Marco, M.D.; Papaleo, E.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Towards mechanistic models for genotype-phenotype correlations in phenylketonuria using protein stability calculations. Hum. Mutat. 2019, 40, 444–457. [Google Scholar] [CrossRef]
- Stein, A.; Fowler, D.M.; Hartmann-Petersen, R.; Lindorff-Larsen, K. Biophysical and Mechanistic Models for Disease-Causing Protein Variants. Trends Biochem. Sci. 2019, 44, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Blouin, J.M.; Bernardo-Seisdedos, G.; Sasso, E.; Esteve, J.; Ged, C.; Lalanne, M.; Sanz-Parra, A.; Urquiza, P.; de Verneuil, H.; Millet, O.; et al. Missense UROS mutations causing congenital erythropoietic porphyria reduce UROS homeostasis that can be rescued by proteasome inhibition. Hum. Mol. Genet. 2017, 26, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Inobe, T.; Matouschek, A. Paradigms of protein degradation by the proteasome. Curr. Opin. Struct. Biol. 2014, 24, 156–164. [Google Scholar] [CrossRef]
- Guharoy, M.; Bhowmick, P.; Sallam, M.; Tompa, P. Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system. Nat. Commun. 2016, 7, 10239. [Google Scholar] [CrossRef] [PubMed]
- Gersing, S.K.; Wang, Y.; Grønbæk-Thygesen, M.; Kampmeyer, C.; Clausen, L.; Andréasson, C.; Stein, A.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Evolutionarily conserved chaperone-mediated proteasomal degradation of a disease-linked aspartoacylase variant. bioRxiv 2020. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Palomino-Morales, R.J.; Fuchs, J.E.; Padin-Gonzalez, E.; Mesa-Torres, N.; Salido, E.; Timson, D.J.; Pey, A.L. Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep. 2016, 6, 20331. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta 2014, 1842, 2163–2173. [Google Scholar] [CrossRef]
- Pey, A.L.; Padin-Gonzalez, E.; Mesa-Torres, N.; Timson, D.J. The metastability of human UDP-galactose 4′-epimerase (GALE) is increased by variants associated with type III galactosemia but decreased by substrate and cofactor binding. Arch. Biochem. Biophys. 2014, 562, 103–114. [Google Scholar] [CrossRef]
- Fuchs, J.E.; Muñoz, I.G.; Timson, D.J.; Pey, A.L. Experimental and computational evidence on conformational fluctuations as a source of catalytic defects in genetic diseases. RSC Adv. 2016, 6, 58604. [Google Scholar] [CrossRef]
- Thomas, D.W.; Burns, J.; Audette, J.; Carroll, A.; Dow-Hygelund, C.; Hay, M. Clinical Development Success Rates 2006–2015. BIO (Biotechnology Innovation Organization). Ind. Anal. 2016, 1, 16. [Google Scholar]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Cleasby, A.; Davies, T.G.; Hall, R.J.; Ludlow, R.F.; Murray, C.W.; Tisi, D.; Jhoti, H. Crystallographic screening using ultra-low-molecular-weight ligands to guide drug design. Drug Discov. Today 2019, 24, 1081–1086. [Google Scholar] [CrossRef]
- Hall, R.J.; Mortenson, P.N.; Murray, C.W. Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 2014, 116, 82–91. [Google Scholar] [CrossRef]
- Holton, J.B. Effects of galactosemia in utero. Eur. J. Pediatr. 1995, 154, S77–S81. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.T.; Gillett, M.; Holton, J.B.; King, G.S.; Pettit, B.R. Evidence for galactosaemia in utero. Lancet 1980, 1, 603. [Google Scholar] [CrossRef]
- Holton, J.B.; Allen, J.T.; Gillett, M.G. Prenatal diagnosis of disorders of galactose metabolism. J. Inherit. Metab. Dis. 1989, 12 (Suppl. S1), 202–206. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).