
NLRP3 Inflammasome Is Involved in Cocaine-Mediated Potentiation on Behavioral Changes in CX3CR1-Deficient Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Animals and Reagents
2.2. Behavioral Tests
2.3. Immunoblots
2.4. Immunofluorescence
2.5. RNA Extraction, Reverse Transcription, and Quantitative Polymerase Chain Reaction (qPCR)
2.6. IL1β ELISA
2.7. Statistics
3. Results
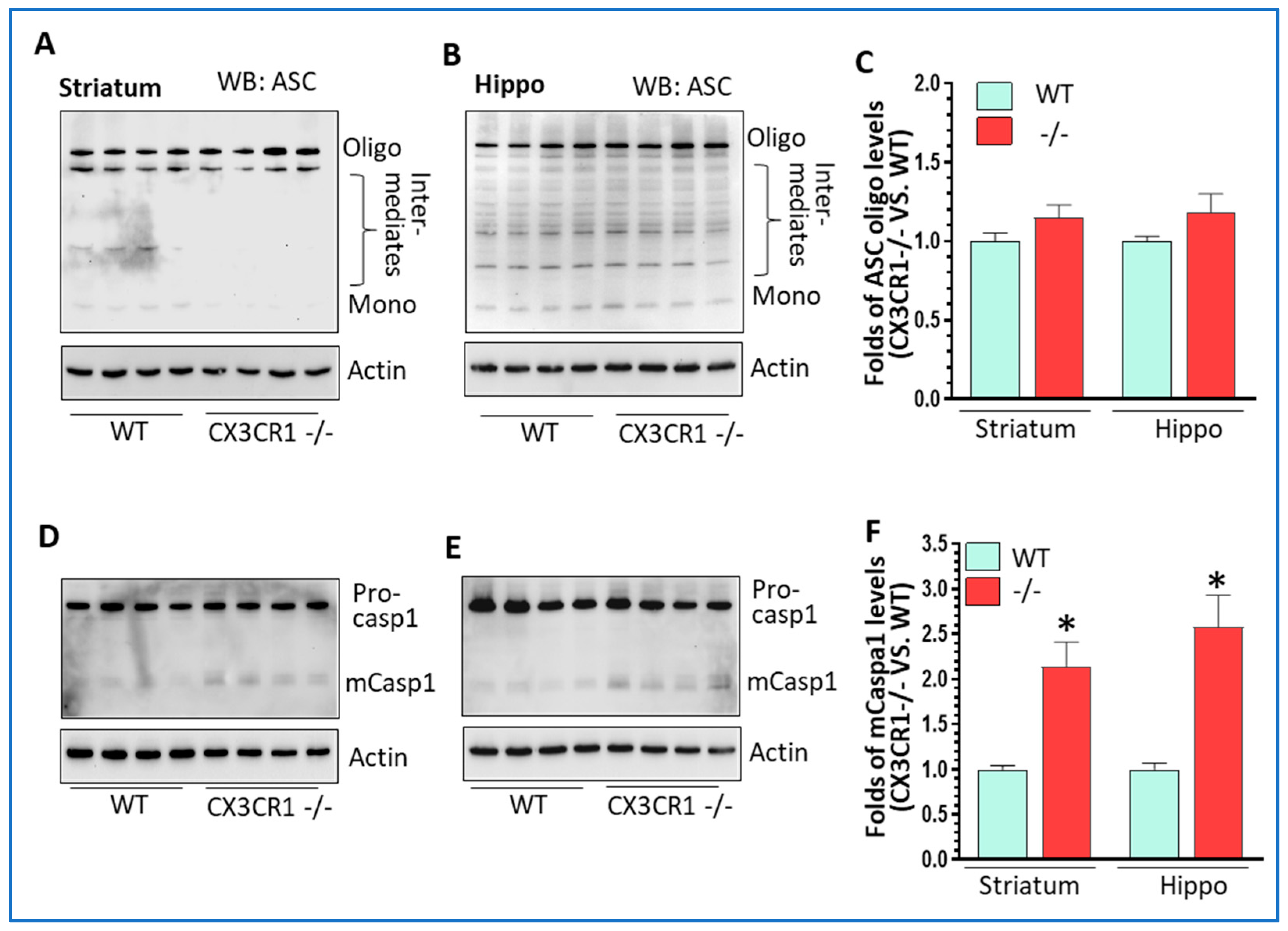
3.1. Enhanced Cocaine-Mediated Locomotor Activity, CPP, and NLRP3 Inflammasome Activity in CX3CR1-Deficient Mice
3.2. CX3CR1 Deficiency-Primed NLRP3 Inflammasome Signaling under Basal Conditions
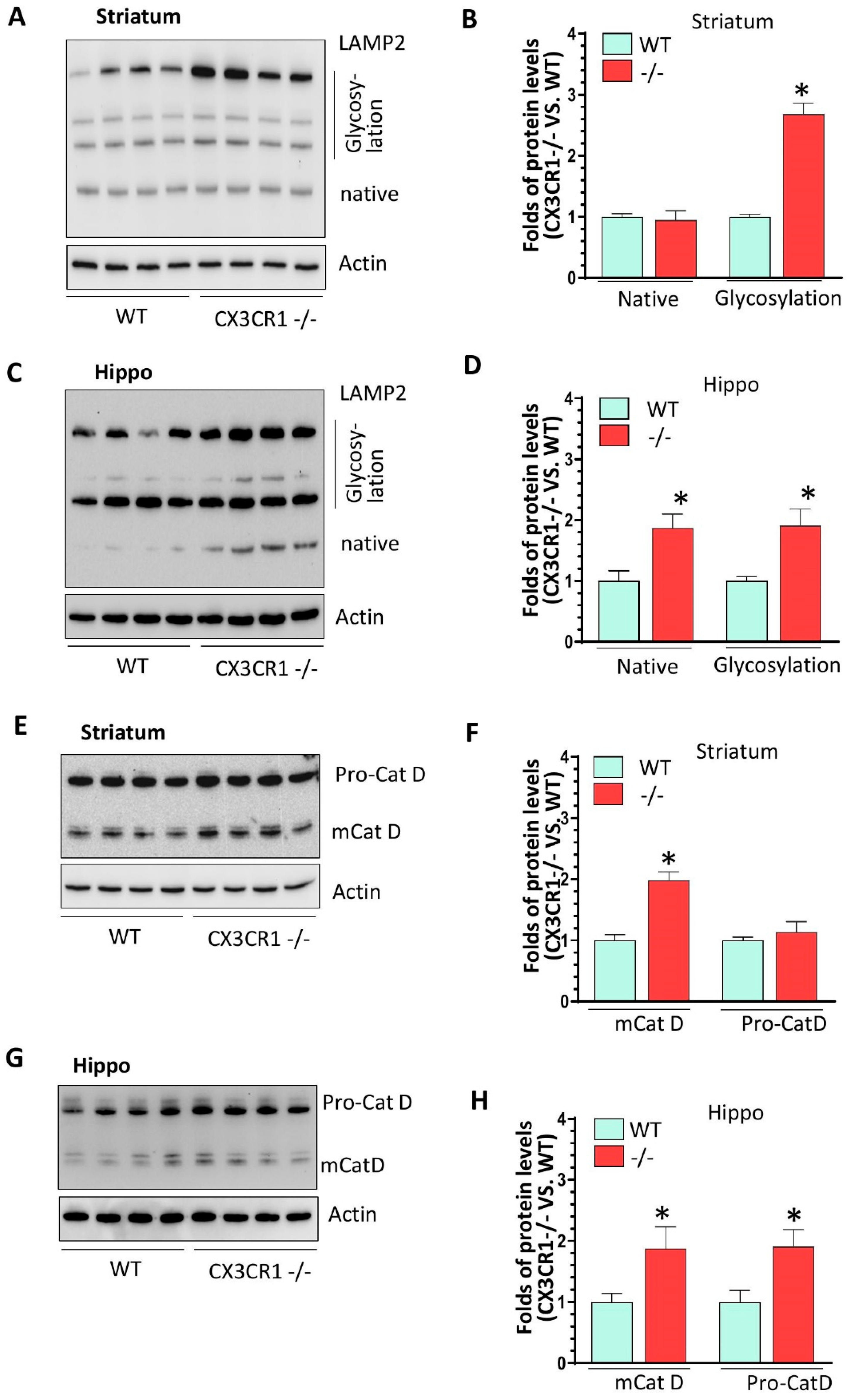
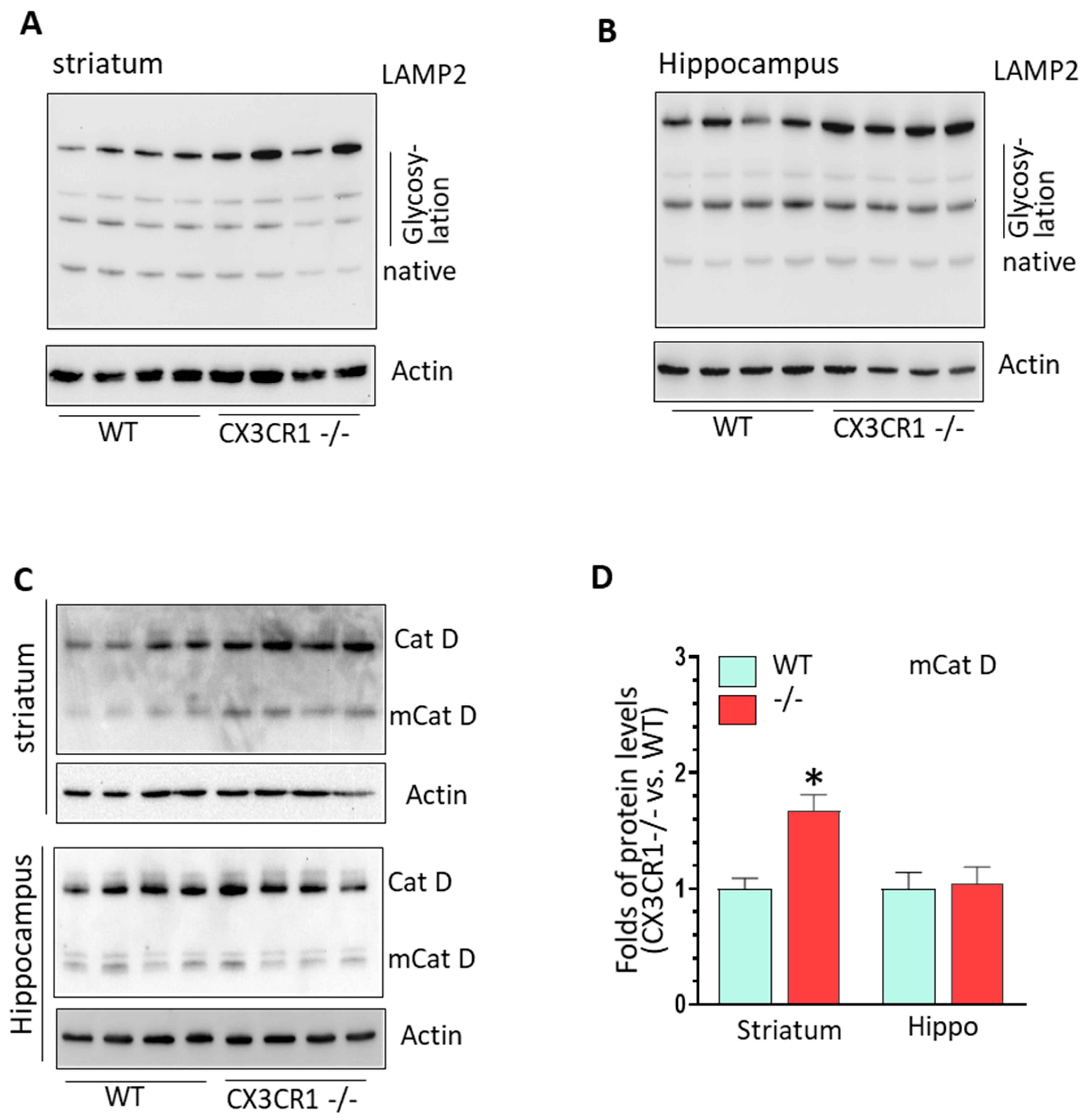
3.3. CX3CR1 Deficiency Increased Lysosome Biogenesis under Basal Condition
4. Discussion
5. Limitation Section
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethics Approval and Consent to Participate
References
- Liao, K.; Guo, M.; Niu, F.; Yang, L.; Callen, S.E.; Buch, S. Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J. Neuroinflammation 2016, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.L.; Liao, K.; Periyasamy, P.; Yang, L.; Cai, Y.; Callen, S.E.; Buch, S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015, 11, 995–1009. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.H.; Wiley, C.A.; Bradberry, C.W. Psychostimulant abuse and neuroinflammation: Emerging evidence of their interconnection. Neurotox. Res. 2013, 23, 174–188. [Google Scholar] [CrossRef]
- Cui, C.; Shurtleff, D.; Harris, R.A. Neuroimmune mechanisms of alcohol and drug addiction. Int. Rev. Neurobiol. 2014, 118, 1–12. [Google Scholar]
- Ahmed, S.H.; Lutjens, R.; van der Stap, L.D.; Lekic, D.; Romano-Spica, V.; Morales, M.; Koob, G.F.; Repunte-Canonigo, V.; Sanna, P.P. Gene expression evidence for remodeling of lateral hypothalamic circuitry in cocaine addiction. Proc. Natl. Acad. Sci. USA 2005, 102, 11533–11538. [Google Scholar] [CrossRef] [Green Version]
- Piechota, M.; Korostynski, M.; Solecki, W.; Gieryk, A.; Slezak, M.; Bilecki, W.; Ziolkowska, B.; Kostrzewa, E.; Cymerman, I.; Swiech, L.; et al. The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol. 2010, 11, R48. [Google Scholar] [CrossRef] [Green Version]
- Northcutt, A.L.; Hutchinson, M.R.; Wang, X.; Baratta, M.V.; Hiranita, T.; Cochran, T.A.; Pomrenze, M.B.; Galer, E.L.; Kopajtic, T.A.; Li, C.M.; et al. DAT isn’t all that: Cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol. Psychiatry 2015, 20, 1525–1537. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Kunitachi, S.; Iyo, M.; Hashimoto, K. The antibiotic minocycline prevents methamphetamine-induced rewarding effects in mice. Pharmacol. Biochem. Behav. 2012, 101, 303–306. [Google Scholar] [CrossRef]
- Chen, H.; Manev, H. Effects of minocycline on cocaine sensitization and phosphorylation of GluR1 receptors in 5-lipoxygenase deficient mice. Neuropharmacology 2011, 60, 1058–1063. [Google Scholar] [CrossRef] [Green Version]
- Kashima, D.T.; Grueter, B.A. Toll-like receptor 4 deficiency alters nucleus accumbens synaptic physiology and drug reward behavior. Proc. Natl. Acad. Sci. USA 2017, 114, 8865–8870. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Bu, Q.; Fu, D.; Shao, X.; Jiang, L.; Guo, W.; Chen, B.; Liu, B.; Hu, Z.; Tian, J.; et al. Toll-like receptor 3 modulates the behavioral effects of cocaine in mice. J. Neuroinflammation 2018, 15, 93. [Google Scholar] [CrossRef]
- Vallender, E.J.; Goswami, D.B.; Shinday, N.M.; Westmoreland, S.V.; Yao, W.D.; Rowlett, J.K. Transcriptomic profiling of the ventral tegmental area and nucleus accumbens in rhesus macaques following long-term cocaine self-administration. Drug Alcohol Depend. 2017, 175, 9–23. [Google Scholar] [CrossRef]
- Little, K.Y.; Ramssen, E.; Welchko, R.; Volberg, V.; Roland, C.J.; Cassin, B. Decreased brain dopamine cell numbers in human cocaine users. Psychiatry Res. 2009, 168, 173–180. [Google Scholar] [CrossRef]
- Kohno, M.; Link, J.; Dennis, L.E.; McCready, H.; Huckans, M.; Hoffman, W.F.; Loftis, J.M. Neuroinflammation in addiction: A review of neuroimaging studies and potential immunotherapies. Pharmacol. Biochem. Behav. 2019, 179, 34–42. [Google Scholar] [CrossRef]
- Lacagnina, M.J.; Rivera, P.D.; Bilbo, S.D. Glial and Neuroimmune Mechanisms as Critical Modulators of Drug Use and Abuse. Neuropsychopharmacology 2017, 42, 156–177. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.; Cooper, M.A.; O’Neill, L.A.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef]
- Cao, S.; Shrestha, S.; Li, J.; Yu, X.; Chen, J.; Yan, F.; Ying, G.; Gu, C.; Wang, L.; Chen, G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep. 2017, 7, 2417. [Google Scholar] [CrossRef] [Green Version]
- Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [Green Version]
- Limatola, C.; Ransohoff, R.M. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell. Neurosci. 2014, 8, 229. [Google Scholar] [CrossRef]
- Slusarczyk, J.; Trojan, E.; Wydra, K.; Glombik, K.; Chamera, K.; Kucharczyk, M.; Budziszewska, B.; Kubera, M.; Lason, W.; Filip, M.; et al. Beneficial impact of intracerebroventricular fractalkine administration on behavioral and biochemical changes induced by prenatal stress in adult rats: Possible role of NLRP3 inflammasome pathway. Biochem. Pharmacol. 2016, 113, 45–56. [Google Scholar] [CrossRef]
- Corona, A.W.; Huang, Y.; O’Connor, J.C.; Dantzer, R.; Kelley, K.W.; Popovich, P.G.; Godbout, J.P. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J. Neuroinflammation 2010, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Robison, A.J.; Vialou, V.; Mazei-Robison, M.; Feng, J.; Kourrich, S.; Collins, M.; Wee, S.; Koob, G.; Turecki, G.; Neve, R.; et al. Behavioral and structural responses to chronic cocaine require a feedforward loop involving DeltaFosB and calcium/calmodulin-dependent protein kinase II in the nucleus accumbens shell. J. Neurosci. 2013, 33, 4295–4307. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Choi, K.H.; Renthal, W.; Tsankova, N.M.; Theobald, D.E.; Truong, H.T.; Russo, S.J.; Laplant, Q.; Sasaki, T.S.; Whistler, K.N.; et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 2005, 48, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Vialou, V.; Feng, J.; Robison, A.J.; Ku, S.M.; Ferguson, D.; Scobie, K.N.; Mazei-Robison, M.S.; Mouzon, E.; Nestler, E.J. Serum response factor and cAMP response element binding protein are both required for cocaine induction of DeltaFosB. J. Neurosci. 2012, 32, 7577–7584. [Google Scholar] [CrossRef] [Green Version]
- Calipari, E.S.; Godino, A.; Peck, E.G.; Salery, M.; Mervosh, N.L.; Landry, J.A.; Russo, S.J.; Hurd, Y.L.; Nestler, E.J.; Kiraly, D.D. Granulocyte-colony stimulating factor controls neural and behavioral plasticity in response to cocaine. Nat. Commun. 2018, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Zenon, F.; Segarra, A.C.; Gonzalez, M.; Melendez, L.M. Cocaine potentiates cathepsin B secretion and neuronal apoptosis from HIV-infected macrophages. J. Neuroimmune Pharmacol. 2014, 9, 703–715. [Google Scholar] [CrossRef] [Green Version]
- Nassogne, M.C.; Lizarraga, C.; N’Kuli, F.; Van Bambeke, F.; Van Binst, R.; Wallemacq, P.; Tulkens, P.M.; Mingeot-Leclercq, M.P.; Levade, T.; Courtoy, P.J. Cocaine induces a mixed lysosomal lipidosis in cultured fibroblasts, by inactivation of acid sphingomyelinase and inhibition of phospholipase A1. Toxicol. Appl. Pharmacol. 2004, 194, 101–110. [Google Scholar] [CrossRef]
- Liu, C.; Hong, K.; Chen, H.; Niu, Y.; Duan, W.; Liu, Y.; Ji, Y.; Deng, B.; Li, Y.; Li, Z.; et al. Evidence for a protective role of the CX3CL1/CX3CR1 axis in a model of amyotrophic lateral sclerosis. Biol. Chem. 2019, 400, 651–661. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef]
- Trageser, K.J.; Sebastian-Valverde, M.; Naughton, S.X.; Pasinetti, G.M. The Innate Immune System and Inflammatory Priming: Potential Mechanistic Factors in Mood Disorders and Gulf War Illness. Front. Psychiatry 2020, 11, 704. [Google Scholar] [CrossRef]
- Lo Iacono, L.; Catale, C.; Martini, A.; Valzania, A.; Viscomi, M.T.; Chiurchiu, V.; Guatteo, E.; Bussone, S.; Perrone, F.; Di Sabato, P.; et al. From Traumatic Childhood to Cocaine Abuse: The Critical Function of the Immune System. Biol. Psychiatry 2018, 84, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Calcia, M.A.; Bonsall, D.R.; Bloomfield, P.S.; Selvaraj, S.; Barichello, T.; Howes, O.D. Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 2016, 233, 1637–1650. [Google Scholar] [CrossRef] [Green Version]
- Martins-Ferreira, R.; Leal, B.; Costa, P.P.; Ballestar, E. Microglial innate memory and epigenetic reprogramming in neurological disorders. Prog. Neurobiol. 2021, 200, 101971. [Google Scholar] [CrossRef]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Liu, Z.; Condello, C.; Schain, A.; Harb, R.; Grutzendler, J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J. Neurosci. 2010, 30, 17091–17101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabel, M.K.; Zhao, L.; Zhang, Y.; Gonzalez, S.R.; Ma, W.; Wang, X.; Fariss, R.N.; Wong, W.T. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 2016, 64, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Al’Absi, M.; Ginty, A.T.; Lovallo, W.R. Neurobiological mechanisms of early life adversity, blunted stress reactivity and risk for addiction. Neuropharmacology 2021, 188, 108519. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L. Stress, sensitive periods, and substance abuse. Neurobiol. Stress 2019, 10, 100140. [Google Scholar] [CrossRef]
- Dutcher, E.G.; Pama, E.A.C.; Lynall, M.E.; Khan, S.; Clatworthy, M.R.; Robbins, T.W.; Bullmore, E.T.; Dalley, J.W. Early-life stress and inflammation: A systematic review of a key experimental approach in rodents. Brain Neurosci. Adv. 2020, 4. [Google Scholar] [CrossRef]
- Cao, P.; Chen, C.; Liu, A.; Shan, Q.; Zhu, X.; Jia, C.; Peng, X.; Zhang, M.; Farzinpour, Z.; Zhou, W.; et al. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 2021, 109, 2573–2589.e9. [Google Scholar] [CrossRef]
- Catale, C.; Bisicchia, E.; Carola, V.; Viscomi, M.T. Early life stress exposure worsens adult remote microglia activation, neuronal death, and functional recovery after focal brain injury. Brain Behav. Immun. 2021, 94, 89–103. [Google Scholar] [CrossRef]
- Saavedra, L.M.; Hernandez-Velazquez, M.G.; Madrigal, S.; Ochoa-Zarzosa, A.; Torner, L. Long-term activation of hippocampal glial cells and altered emotional behavior in male and female adult rats after different neonatal stressors. Psychoneuroendocrinology 2021, 126, 105164. [Google Scholar] [CrossRef]
- Lei, Y.; Chen, C.J.; Yan, X.X.; Li, Z.; Deng, X.H. Early-life lipopolysaccharide exposure potentiates forebrain expression of NLRP3 inflammasome proteins and anxiety-like behavior in adolescent rats. Brain Res. 2017, 1671, 43–54. [Google Scholar] [CrossRef]
- Niu, L.; Luo, S.S.; Xu, Y.; Wang, Z.; Luo, D.; Yang, H.; Li, W.; He, J.; Zhong, X.L.; Liu, Z.H.; et al. The critical role of the hippocampal NLRP3 inflammasome in social isolation-induced cognitive impairment in male mice. Neurobiol. Learn. Mem. 2020, 175, 107301. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rivera, N.M.; Ortiz-Lopez, L.; Granados-Juarez, A.; Estrada-Camarena, E.M.; Ramirez-Rodriguez, G.B. Melatonin Reverses the Depression-associated Behaviour and Regulates Microglia, Fractalkine Expression and Neurogenesis in Adult Mice Exposed to Chronic Mild Stress. Neuroscience 2020, 440, 316–336. [Google Scholar] [CrossRef] [PubMed]
- Schubert, I.; Ahlbrand, R.; Winter, A.; Vollmer, L.; Lewkowich, I.; Sah, R. Enhanced fear and altered neuronal activation in forebrain limbic regions of CX3CR1-deficient mice. Brain Behav. Immun. 2018, 68, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Ke, X.; Liu, Q.; Fu, Q.; Majnik, A.; Lane, R. Adverse early life environment increases hippocampal microglia abundance in conjunction with decreased neural stem cells in juvenile mice. Int. J. Dev. Neurosci. 2016, 55, 56–65. [Google Scholar] [CrossRef]
- Gracia-Rubio, I.; Moscoso-Castro, M.; Pozo, O.J.; Marcos, J.; Nadal, R.; Valverde, O. Maternal separation induces neuroinflammation and long-lasting emotional alterations in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 104–117. [Google Scholar] [CrossRef]
- Zhou, L.; Wu, Z.; Wang, G.; Xiao, L.; Wang, H.; Sun, L.; Xie, Y. Long-term maternal separation potentiates depressive-like behaviours and neuroinflammation in adult male C57/BL6J mice. Pharmacol. Biochem. Behav. 2020, 196, 172953. [Google Scholar] [CrossRef]
- Mi, J.; Yang, Y.; Yao, H.; Huan, Z.; Xu, C.; Ren, Z.; Li, W.; Tang, Y.; Fu, R.; Ge, X. Inhibition of heat shock protein family A member 8 attenuates spinal cord ischemia-reperfusion injury via astrocyte NF-kappaB/NLRP3 inflammasome pathway: HSPA8 inhibition protects spinal ischemia-reperfusion injury. J. Neuroinflammation 2021, 18, 170. [Google Scholar] [CrossRef]
- Hong, Y.; Liu, Y.; Yu, D.; Wang, M.; Hou, Y. The neuroprotection of progesterone against Abeta-induced NLRP3-Caspase-1 inflammasome activation via enhancing autophagy in astrocytes. Int. Immunopharmacol. 2019, 74, 105669. [Google Scholar] [CrossRef]
- Araos, P.; Pedraz, M.; Serrano, A.; Lucena, M.; Barrios, V.; Garcia-Marchena, N.; Campos-Cloute, R.; Ruiz, J.J.; Romero, P.; Suarez, J.; et al. Plasma profile of pro-inflammatory cytokines and chemokines in cocaine users under outpatient treatment: Influence of cocaine symptom severity and psychiatric co-morbidity. Addict. Biol. 2015, 20, 756–772. [Google Scholar] [CrossRef]
- Eagle, A.L.; Singh, R.; Kohler, R.J.; Friedman, A.L.; Liebowitz, C.P.; Galloway, M.P.; Enman, N.M.; Jutkiewicz, E.M.; Perrine, S.A. Single prolonged stress effects on sensitization to cocaine and cocaine self-administration in rats. Behav. Brain Res. 2015, 284, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Burke, A.R.; Miczek, K.A. Stress in adolescence and drugs of abuse in rodent models: Role of dopamine, CRF, and HPA axis. Psychopharmacology 2014, 231, 1557–1580. [Google Scholar] [CrossRef] [Green Version]
- Merkel, S.F.; Razmpour, R.; Lutton, E.M.; Tallarida, C.S.; Heldt, N.A.; Cannella, L.A.; Persidsky, Y.; Rawls, S.M.; Ramirez, S.H. Adolescent Traumatic Brain Injury Induces Chronic Mesolimbic Neuroinflammation with Concurrent Enhancement in the Rewarding Effects of Cocaine in Mice during Adulthood. J. Neurotrauma 2017, 34, 165–181. [Google Scholar] [CrossRef] [Green Version]
- Scheidell, J.D.; Quinn, K.; McGorray, S.P.; Frueh, B.C.; Beharie, N.N.; Cottler, L.B.; Khan, M.R. Childhood Traumatic Experiences and the Association with Marijuana and Cocaine Use in Adolescence through Adulthood. Addiction 2017, 113, 44–56. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.-L.; Chivero, E.T.; Callen, S.E.; Buch, S. NLRP3 Inflammasome Is Involved in Cocaine-Mediated Potentiation on Behavioral Changes in CX3CR1-Deficient Mice. J. Pers. Med. 2021, 11, 963. https://doi.org/10.3390/jpm11100963
Guo M-L, Chivero ET, Callen SE, Buch S. NLRP3 Inflammasome Is Involved in Cocaine-Mediated Potentiation on Behavioral Changes in CX3CR1-Deficient Mice. Journal of Personalized Medicine. 2021; 11(10):963. https://doi.org/10.3390/jpm11100963
Chicago/Turabian StyleGuo, Ming-Lei, Ernest T. Chivero, Shannon E. Callen, and Shilpa Buch. 2021. "NLRP3 Inflammasome Is Involved in Cocaine-Mediated Potentiation on Behavioral Changes in CX3CR1-Deficient Mice" Journal of Personalized Medicine 11, no. 10: 963. https://doi.org/10.3390/jpm11100963
APA StyleGuo, M.-L., Chivero, E. T., Callen, S. E., & Buch, S. (2021). NLRP3 Inflammasome Is Involved in Cocaine-Mediated Potentiation on Behavioral Changes in CX3CR1-Deficient Mice. Journal of Personalized Medicine, 11(10), 963. https://doi.org/10.3390/jpm11100963