Genomic Screening Identifies Individuals at High Risk for Hereditary Transthyretin Amyloidosis

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Setting and Population
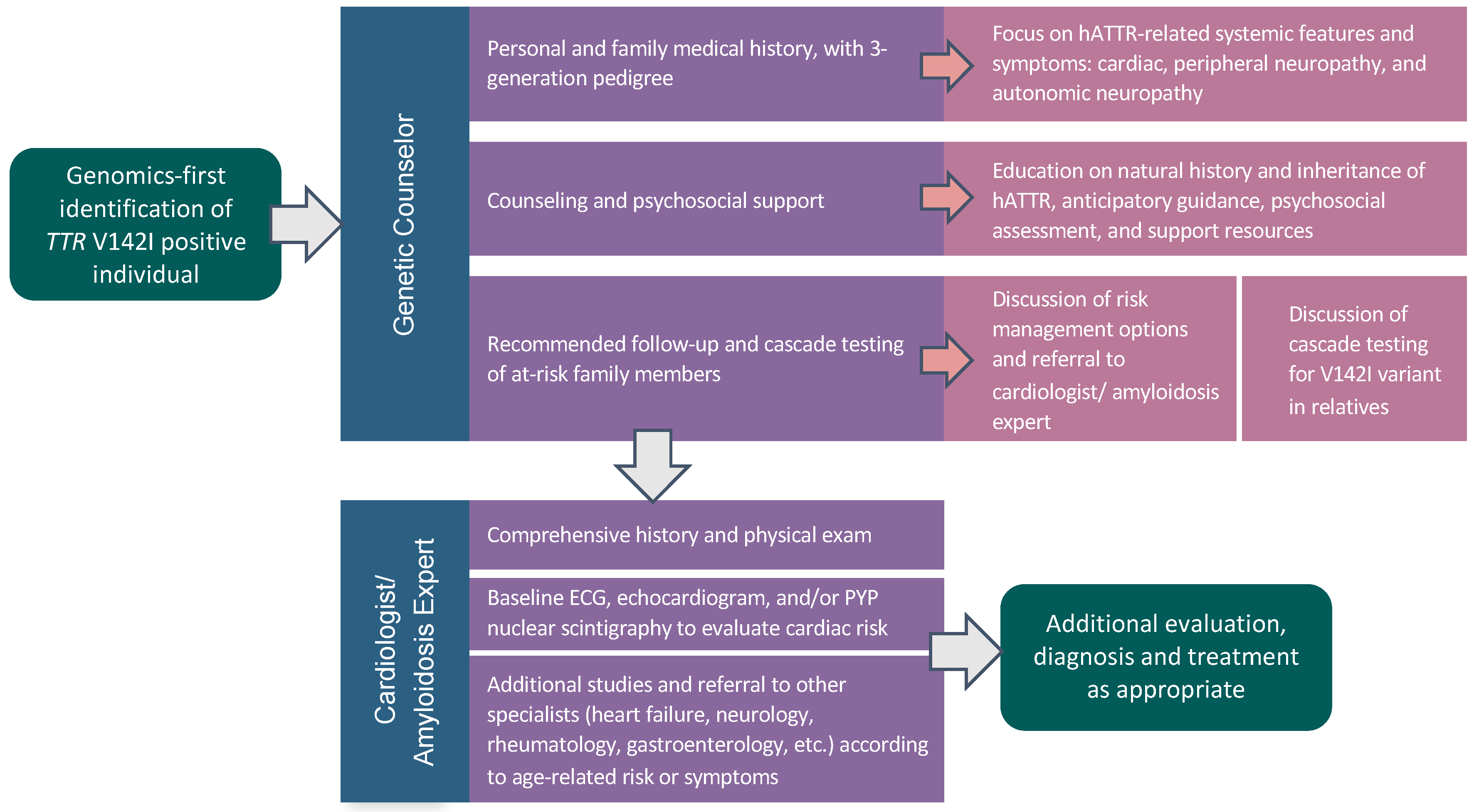
2.2. Pilot Genomic Screening Program
2.3. TTR V142I Results Disclosure
2.4. Characteristics of V142I-Positive Individuals at Result Disclosure
2.5. Post-Result Disclosure Follow-Up and Outcomes
2.6. Statistical Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, M.F.; Giovanni, M.A. Bringing monogenic disease screening to the clinic. Nat. Med. 2020, 26, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, A.H.; Lester Kirchner, H.; Schwartz, M.L.B.; Kelly, M.A.; Schmidlen, T.; Jones, L.K.; Hallquist, M.L.G.; Rocha, H.; Betts, M.; Schwiter, R.; et al. Clinical outcomes of a genomic screening program for actionable genetic conditions. Genet. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Grzymski, J.J.; Elhanan, G.; Morales Rosado, J.A.; Smith, E.; Schlauch, K.A.; Read, R.; Rowan, C.; Slotnick, N.; Dabe, S.; Metcalf, W.J.; et al. Population genetic screening efficiently identifies carriers of autosomal dominant diseases. Nat. Med. 2020, 26, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Lemke, A.A.; Amendola, L.M.; Kuchta, K.; Dunnenberger, H.M.; Thompson, J.; Johnson, C.; Ilbawi, N.; Oshman, L.; Hulick, P.J. Primary Care Physician Experiences with Integrated Population-Scale Genetic Testing: A Mixed-Methods Assessment. J Pers. Med. 2020, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Soper, E.R.; Odgis, J.A.; Cullina, S.; Bobo, D.; Moscati, A.; Rodriguez, J.E.; Team, C.G.; Regeneron Genetics, C.; Loos, R.J.F.; et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 2019, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Manickam, K.; Jones, L.K.; Wright, E.A.; Hartzel, D.N.; Gonzaga-Jauregui, C.; O’Dushlaine, C.; Leader, J.B.; Lester Kirchner, H.; Lindbuchler, D.a.M.; et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016, 354. [Google Scholar] [CrossRef]
- Manickam, K.; Buchanan, A.H.; Schwartz, M.L.B.; Hallquist, M.L.G.; Williams, J.L.; Rahm, A.K.; Rocha, H.; Savatt, J.M.; Evans, A.E.; Butry, L.M.; et al. Exome sequencing-based screening for BRCA1/2 expected pathogenic variants among adult biobank participants. JAMA Netw. Open 2018, 1, e182140. [Google Scholar] [CrossRef]
- Buchanan, A.H.; Manickam, K.; Meyer, M.N.; Wagner, J.K.; Hallquist, M.L.G.; Williams, J.L.; Rahm, A.K.; Williams, M.S.; Chen, Z.-M.E.; Shah, C.K.; et al. Early cancer diagnoses through BRCA1/2 screening of unselected adult biobank participants. Genet. Med. 2018, 20, 554–558. [Google Scholar] [CrossRef]
- Rowczenio, D.M.; Noor, I.; Gillmore, J.D.; Lachmann, H.J.; Whelan, C.; Hawkins, P.N.; Obici, L.; Westermark, P.; Grateau, G.; Wechalekar, A.D. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum. Mutat. 2014, 35, E2403–E2412. [Google Scholar] [CrossRef]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2001. [Google Scholar]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef]
- Hawkins, P.N.; Ando, Y.; Dispenzeri, A.; Gonzalez-Duarte, A.; Adams, D.; Suhr, O.B. Evolving landscape in the management of transthyretin amyloidosis. Ann. Med. 2015, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Kumar Dharmarajan, M.S.M. Transthyretin cardiac amyloidoses in older North Americans. J. Am. Geriatr. Soc. 2012, 60, 765. [Google Scholar] [CrossRef] [PubMed]
- Lahuerta Pueyo, C.; Aibar Arregui, M.Á.; Gracia Gutierrez, A.; Bueno Juana, E.; Menao Guillén, S. Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur. J. Hum. Genet. 2019, 27, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.R.; Alexander, A.A.; Tagoe, C.; Garvey, W.T.; Williams, S.M.; Tishkoff, S.; Modiano, D.; Sirima, S.B.; Kalidi, I.; Toure, A.; et al. The prevalence and distribution of the amyloidogenic transthyretin (TTR) V122I allele in Africa. Mol. Genet. Genomic. Med. 2016, 4, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Hamidi Asl, K.; Yazaki, M.; Benson, M.D. A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid 2005, 12, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Damrauer, S.M.; Chaudhary, K.; Cho, J.H.; Liang, L.W.; Argulian, E.; Chan, L.; Dobbyn, A.; Guerraty, M.A.; Judy, R.; Kay, J.; et al. Association of the V122I Hereditary Transthyretin Amyloidosis Genetic Variant With Heart Failure Among Individuals of African or Hispanic/Latino Ancestry. JAMA 2019, 322, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.R.; Pastore, R.D.; Yaghoubian, R.; Kane, I.; Gallo, G.; Buck, F.S.; Buxbaum, J.N. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N. Engl. J. Med. 1997, 336, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.C.; Buxbaum, J.N.; Shah, A.M.; Falk, R.H.; Claggett, B.; Kitzman, D.W.; Mosley, T.H.; Butler, K.R.; Boerwinkle, E.; Solomon, S.D. The amyloidogenic V122I transthyretin variant in elderly black Americans. N. Engl. J. Med. 2015, 372, 21–29. [Google Scholar] [CrossRef]
- Carr, A.S.; Pelayo-Negro, A.L.; Jaunmuktane, Z.; Scalco, R.S.; Hutt, D.; Evans, M.R.B.; Heally, E.; Brandner, S.; Holton, J.; Blake, J.; et al. Transthyretin V122I amyloidosis with clinical and histological evidence of amyloid neuropathy and myopathy. Neuromuscul. Disord. 2015, 25, 511–515. [Google Scholar] [CrossRef]
- Maurer, M.S.; Hanna, M.; Grogan, M.; Dispenzieri, A.; Witteles, R.; Drachman, B.; Judge, D.P.; Lenihan, D.J.; Gottlieb, S.S.; Shah, S.J.; et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J. Am. Coll. Cardiol. 2016, 68, 161–172. [Google Scholar] [CrossRef]
- Yanagisawa, A.; Ueda, M.; Sueyoshi, T.; Okada, T.; Fujimoto, T.; Ogi, Y.; Kitagawa, K.; Tasaki, M.; Misumi, Y.; Oshima, T.; et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod. Pathol. 2015, 28, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Aus dem Siepen, F.; Hein, S.; Prestel, S.; Baumgärtner, C.; Schönland, S.; Hegenbart, U.; Röcken, C.; Katus, H.A.; Kristen, A.V. Carpal tunnel syndrome and spinal canal stenosis: Harbingers of transthyretin amyloid cardiomyopathy? Clin. Res. Cardiol. 2019, 108, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sekijima, Y.; Yazaki, M.; Tojo, K.; Yoshinaga, T.; Doden, T.; Koyama, J.; Yanagisawa, S.; Ikeda, S.-I. Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Sperry, B.W.; Reyes, B.A.; Ikram, A.; Donnelly, J.P.; Phelan, D.; Jaber, W.A.; Shapiro, D.; Evans, P.J.; Maschke, S.; Kilpatrick, S.E.; et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J. Am. Coll. Cardiol. 2018, 72, 2040–2050. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Office of the Commissioner. FDA Approves New Treatments for Heart Disease Caused by A Serious Rare Disease, Transthyretin Mediated Amyloidosis. FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatments-heart-disease-caused-serious-rare-disease-transthyretin-mediated#:~:text=On%20May%203%2C%20the%20U.S.,approved%20treatments%20for%20ATTR%2DCM (accessed on 20 December 2020).
- Abul-Husn, N.S.; Soper, E.R.; Braganza, G.T.; Rodriguez, J.E.; Zeid, N.; Cullina, S.; Bobo, D.; Moscati, A.; Merkelson, A.; Loos, R.J.F.; et al. Implementing genomic screening in diverse populations. medRxiv 2021. [Google Scholar] [CrossRef]
- Belbin, G.M.; Wenric, S.; Cullina, S.; Glicksberg, B.S.; Moscati, A.; Wojcik, G.L.; Shemirani, R.; Beckmann, N.D.; Cohain, A.; Sorokin, E.P.; et al. Towards a fine-scale population health monitoring system. bioRxiv 2019. [Google Scholar] [CrossRef]
- Tier 1 Genomics Applications and their Importance to Public Health | CDC. Available online: https://www.cdc.gov/genomics/implementation/toolkit/tier1.htm (accessed on 20 December 2020).
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Sema4: A Patient-Centered Health Intelligence Company. Available online: https://sema4.com/ (accessed on 20 December 2020).
- Gertz, M.A.; Mauermann, M.L.; Grogan, M.; Coelho, T. Advances in the treatment of hereditary transthyretin amyloidosis: A review. Brain Behav. 2019, 9, e01371. [Google Scholar] [CrossRef] [PubMed]
- Conceição, I.; González-Duarte, A.; Obici, L.; Schmidt, H.H.J.; Simoneau, D.; Ong, M.-L.; Amass, L. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J. Peripher. Nerv. Syst. 2016, 21, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.; Alexander, A.; Koziol, J.; Tagoe, C.; Fox, E.; Kitzman, D. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am. Heart J. 2010, 159, 864–870. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ruberg, F.L. Transthyretin V122I (pV142I)* cardiac amyloidosis: An age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet. Med. 2017, 19, 733–742. [Google Scholar] [CrossRef]
- Dungu, J.N.; Papadopoulou, S.A.; Wykes, K.; Mahmood, I.; Marshall, J.; Valencia, O.; Fontana, M.; Whelan, C.J.; Gillmore, J.D.; Hawkins, P.N.; et al. Afro-Caribbean heart failure in the United Kingdom: Cause, outcomes, and ATTR V122I cardiac amyloidosis. Circ. Heart Fail. 2016, 9. [Google Scholar] [CrossRef]
- Connors, L.H.; Prokaeva, T.; Lim, A.; Théberge, R.; Falk, R.H.; Doros, G.; Berg, A.; Costello, C.E.; O’Hara, C.; Seldin, D.C.; et al. Cardiac amyloidosis in African Americans: Comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am. Heart J. 2009, 158, 607–614. [Google Scholar] [CrossRef]
- Gopal, D.M.; Ruberg, F.L.; Siddiqi, O.K. Impact of genetic testing in transthyretin (ATTR) cardiac amyloidosis. Curr. Heart Fail. Rep. 2019, 16, 180–188. [Google Scholar] [CrossRef]
- Grandis, M.; Obici, L.; Luigetti, M.; Briani, C.; Benedicenti, F.; Bisogni, G.; Canepa, M.; Cappelli, F.; Danesino, C.; Fabrizi, G.M.; et al. Recommendations for pre-symptomatic genetic testing for hereditary transthyretin amyloidosis in the era of effective therapy: A multicenter Italian consensus. Orphanet. J. Rare Dis. 2020, 15, 348. [Google Scholar] [CrossRef]
- 23andMe. A New 23andMe Genetic Health Risk Report Brings to Light Underdiagnosed Condition—23andMe Blog. Available online: https://blog.23andme.com/health-traits/a-new-23andme-genetic-health-risk-report-brings-to-light-underdiagnosed-condition/ (accessed on 20 December 2020).
- Conceição, I.; Damy, T.; Romero, M.; Galán, L.; Attarian, S.; Luigetti, M.; Sadeh, M.; Sarafov, S.; Tournev, I.; Ueda, M. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid 2019, 26, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Lorenzini, M.; Longhi, S.; Milandri, A.; Gagliardi, C.; Bartolomei, I.; Salvi, F.; Maurer, M.S. Cardiac amyloidosis: The great pretender. Heart Fail. Rev. 2015, 20, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The missing diversity in human genetic studies. Cell 2019, 177, 26–31. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Characteristics | Participants (N = 32) |
|---|---|
| Age, median (range) | 57 (30–79) |
| Female, No. (%) | 26 (81) |
| Self-reported race/ethnicity, No. (%) | |
| African American/African | 17 (53) |
| Hispanic/Latinx | 15 (47) |
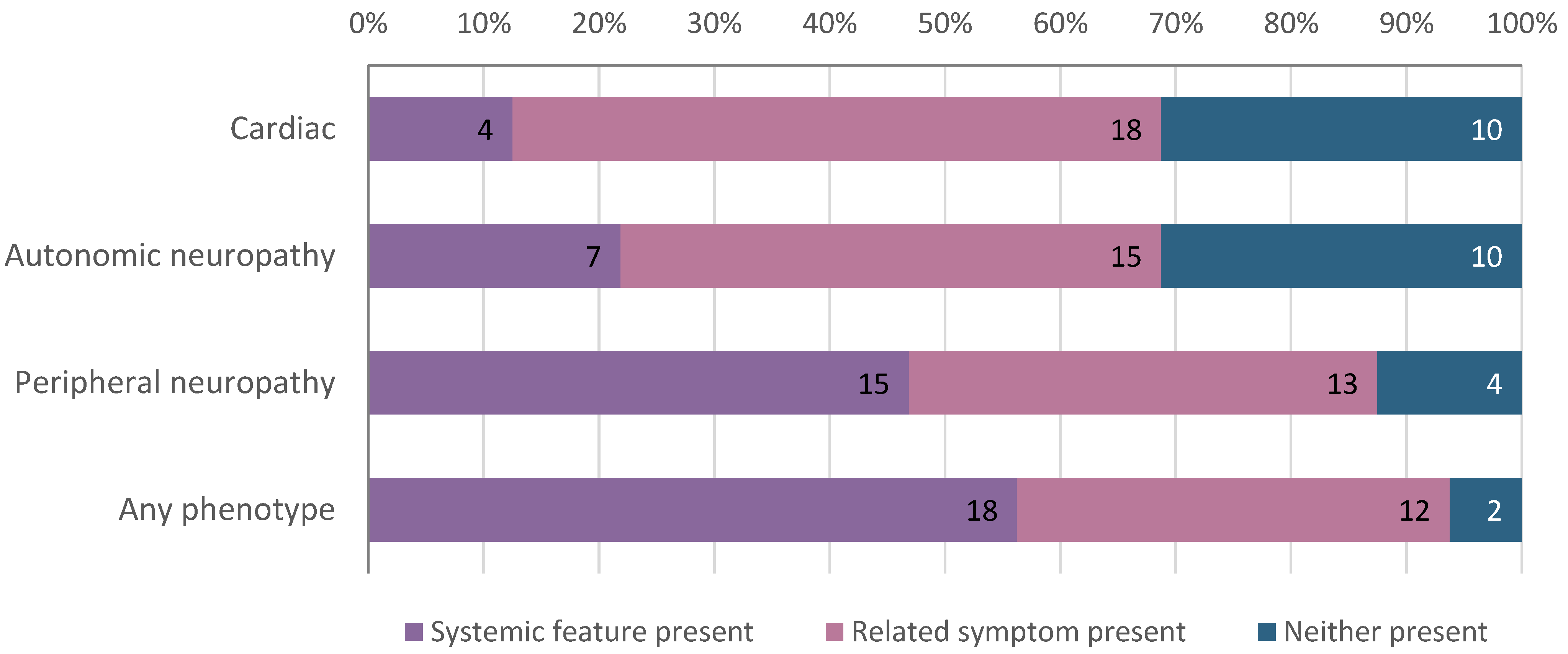
| Personal history of hATTR-related feature, No. (%) | 18 (56) |
| Family history of hATTR-related feature, No. (%) | 15 (47) |
| Follow-Up with Specialists (N = 32) | No. (%) | Weeks Post-Results Disclosure, Median (Range) |
|---|---|---|
| Cardiologist/Cardiovascular geneticist | 18 (56) | 4.9 (0.4–34.9) |
| Heart failure specialist | 2 (6) | 15.4 (1.9–28.9) |
| Neurologist | 2 (6) | 27.4 (16.0–38.7) |
| Interventions among Individuals Seen by a Specialist (N = 16) | No.* (%) | Weeks Post-Results Disclosure, Median (Range) |
| ECG | 10 of 12 (83) | 3.0 (0.4–20.0) |
| Echocardiogram | 12 of 16 (75) | 13.5 (2.1–41.0) |
| Tc-99m-PYP scintigraphy | 10 of 13 (77) | 8.3 (2.1–38.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soper, E.R.; Suckiel, S.A.; Braganza, G.T.; Kontorovich, A.R.; Kenny, E.E.; Abul-Husn, N.S. Genomic Screening Identifies Individuals at High Risk for Hereditary Transthyretin Amyloidosis. J. Pers. Med. 2021, 11, 49. https://doi.org/10.3390/jpm11010049
Soper ER, Suckiel SA, Braganza GT, Kontorovich AR, Kenny EE, Abul-Husn NS. Genomic Screening Identifies Individuals at High Risk for Hereditary Transthyretin Amyloidosis. Journal of Personalized Medicine. 2021; 11(1):49. https://doi.org/10.3390/jpm11010049
Chicago/Turabian StyleSoper, Emily R., Sabrina A. Suckiel, Giovanna T. Braganza, Amy R. Kontorovich, Eimear E. Kenny, and Noura S. Abul-Husn. 2021. "Genomic Screening Identifies Individuals at High Risk for Hereditary Transthyretin Amyloidosis" Journal of Personalized Medicine 11, no. 1: 49. https://doi.org/10.3390/jpm11010049
APA StyleSoper, E. R., Suckiel, S. A., Braganza, G. T., Kontorovich, A. R., Kenny, E. E., & Abul-Husn, N. S. (2021). Genomic Screening Identifies Individuals at High Risk for Hereditary Transthyretin Amyloidosis. Journal of Personalized Medicine, 11(1), 49. https://doi.org/10.3390/jpm11010049