A Conformation Variant of p53 Combined with Machine Learning Identifies Alzheimer Disease in Preclinical and Prodromal Stages

 , , ,
, , ,  , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Participants and Clinical Phenotyping
2.2. Sample Size Considerations for InveCe.Ab
2.3. Immunoassay for Plasma U-p532D3A8+ Biomarker Analysis
2.4. Statistical Analysis
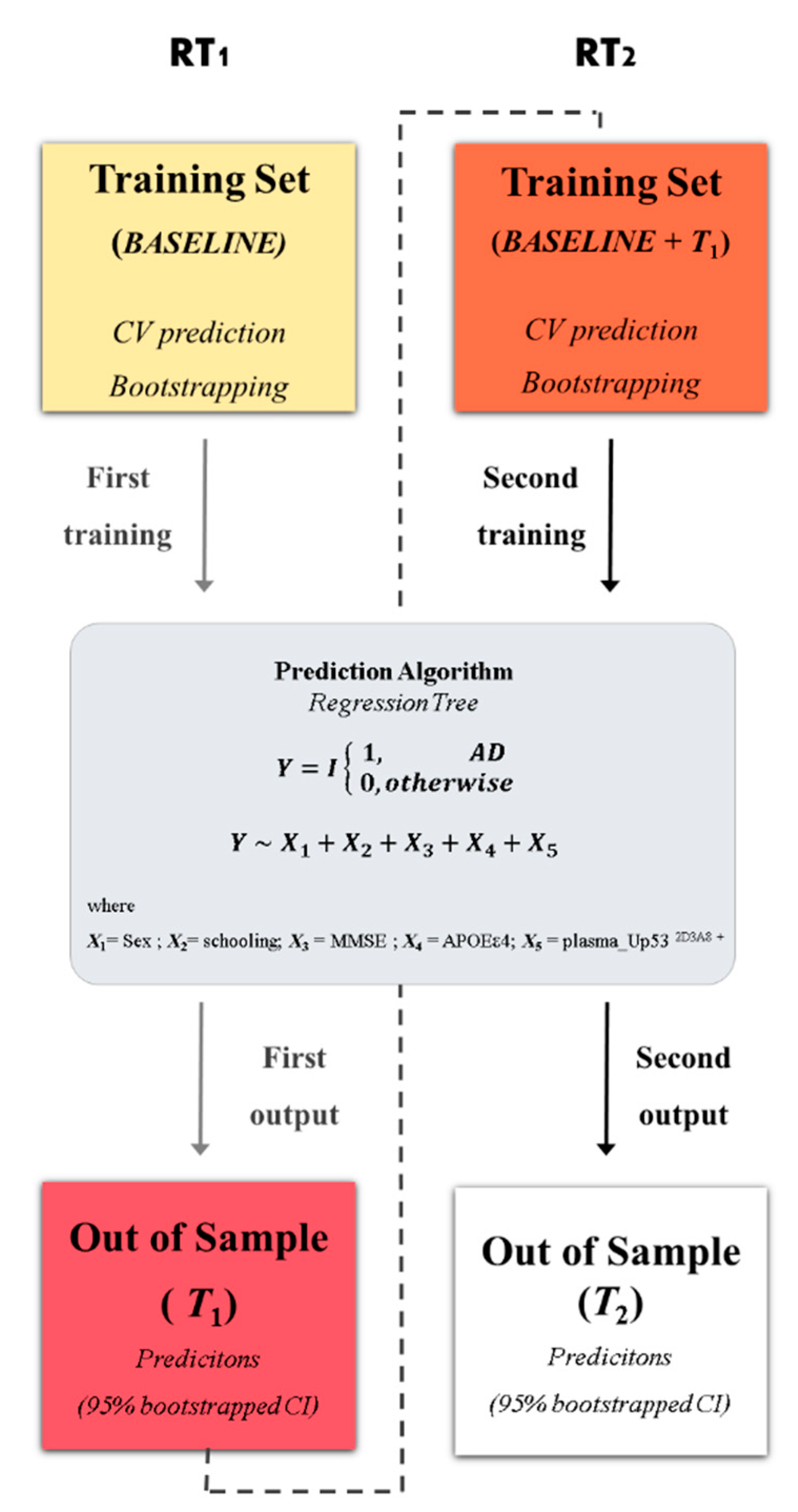
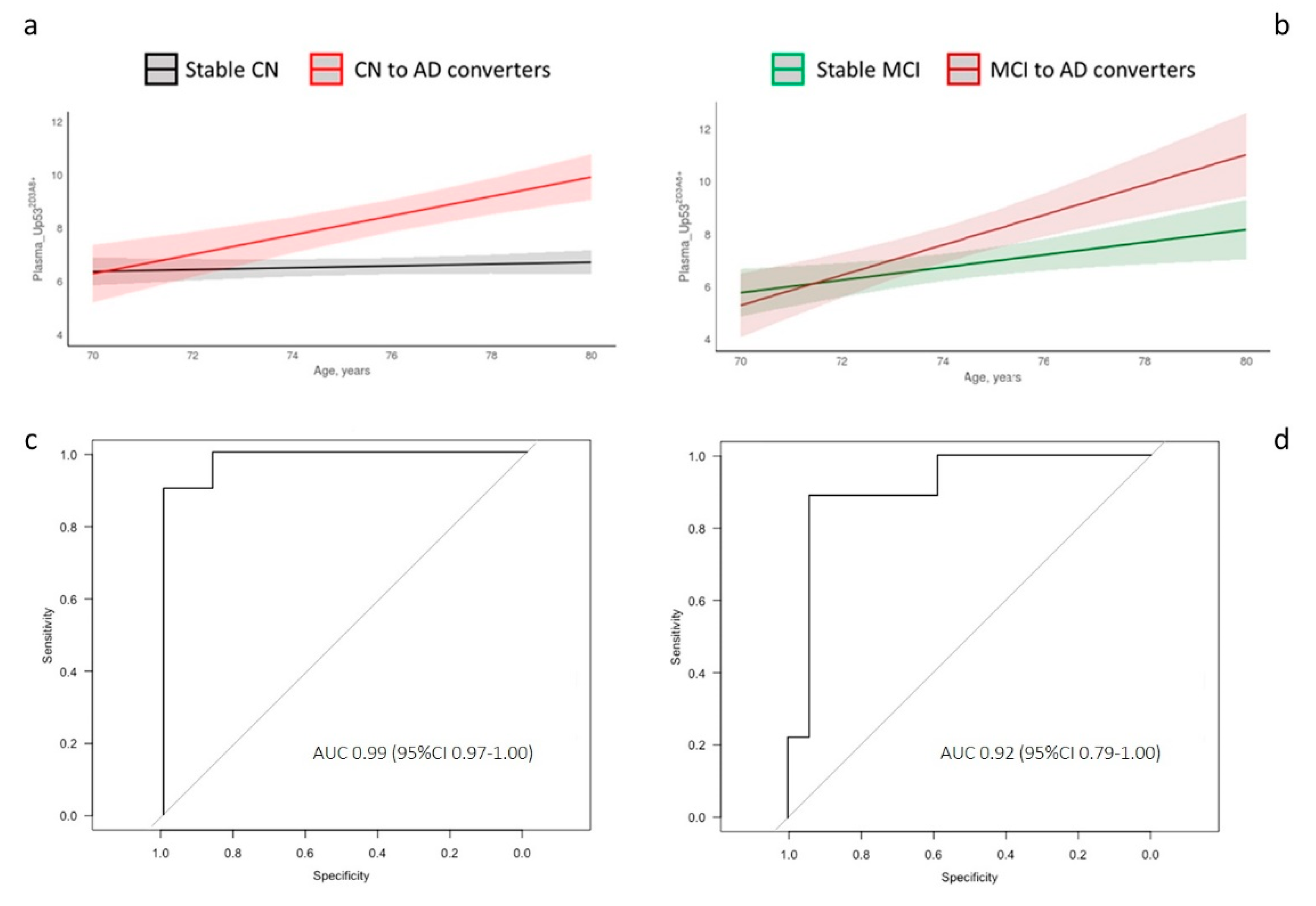
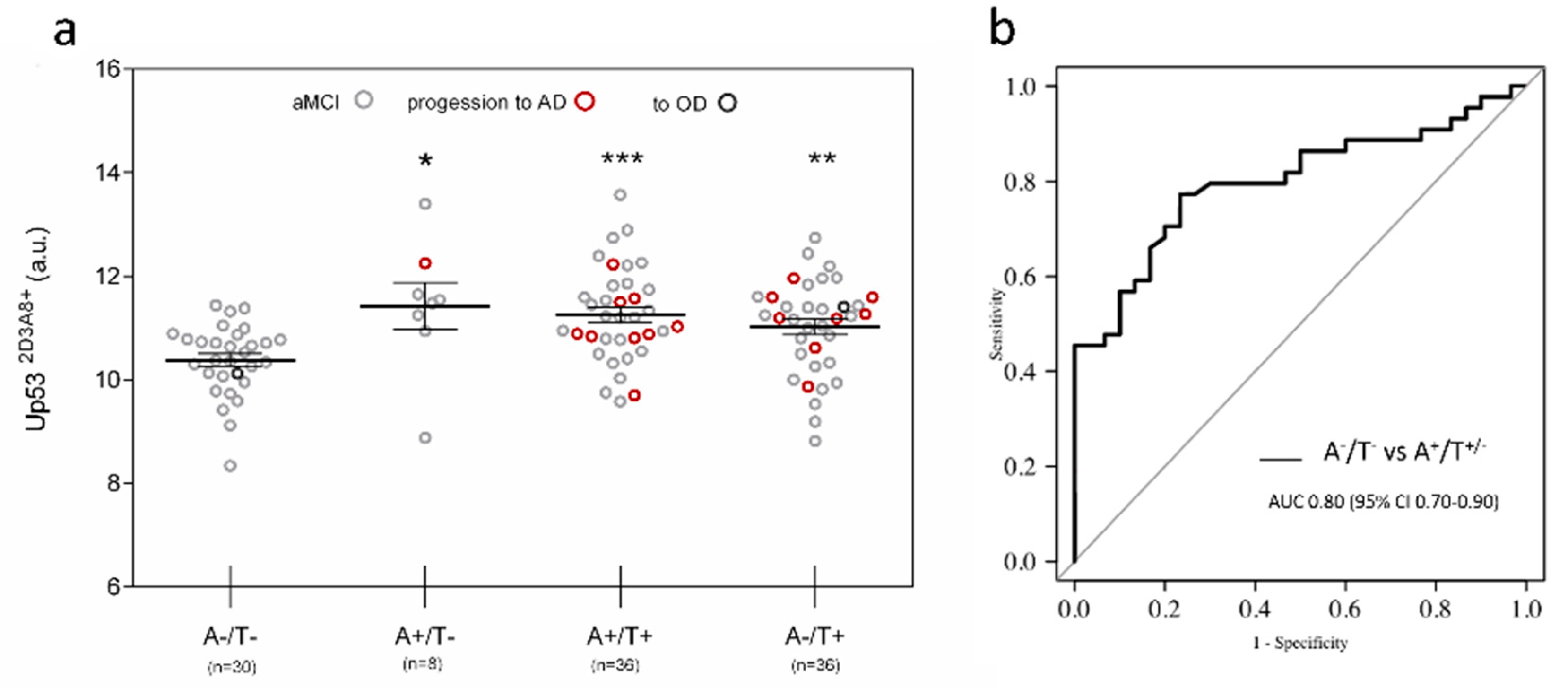
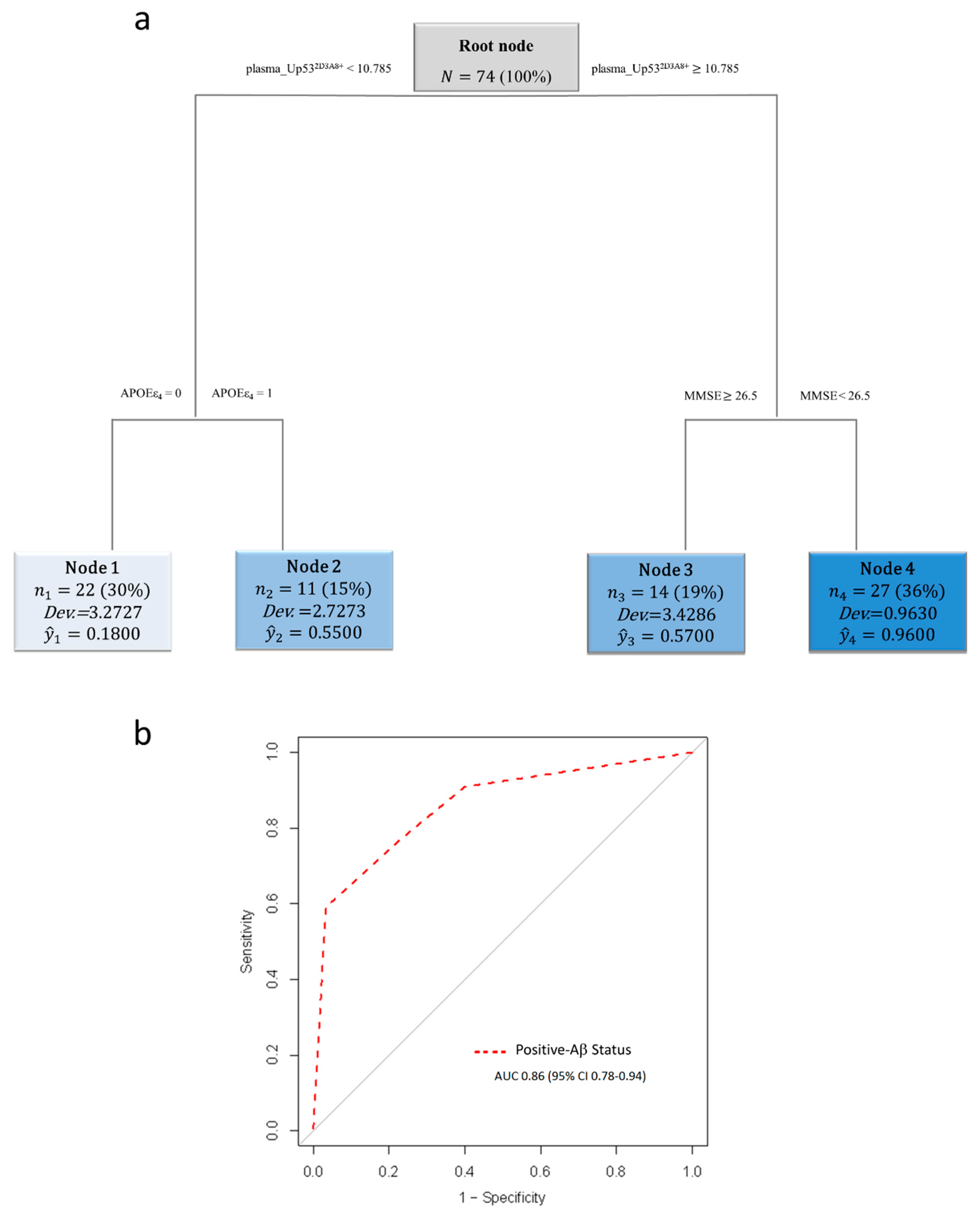
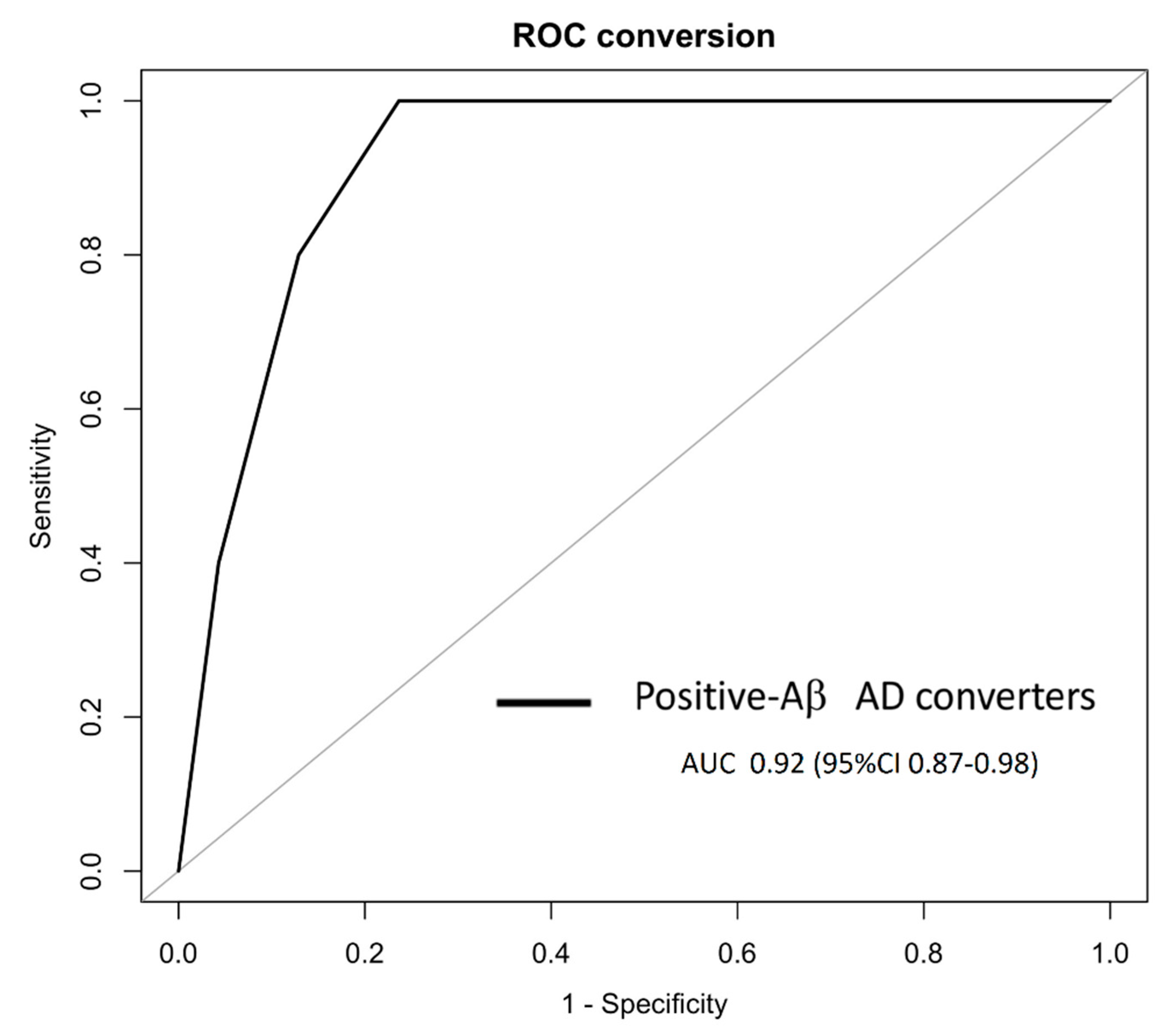
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patterson, C. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018; pp. 32–36. [Google Scholar]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2017. Alzheimer’s Dement. 2017, 3, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Albert, M.L. Amnestic MCI or prodromal Alzheimer’s disease? Lancet Neurol. 2004, 3, 246–248. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A systemic view of Alzheimer disease—insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 703. [Google Scholar] [CrossRef]
- Tramutola, A.; Abate, G.; Lanzillotta, C.; Triani, F.; Barone, E.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Marziano, M.; Memo, M.; et al. Protein nitration profile of CD3(+) lymphocytes from Alzheimer disease patients: Novel hints on immunosenescence and biomarker detection. Free Radic. Biol. Med. 2018, 129, 430–439. [Google Scholar] [CrossRef]
- Jack, C.R.J.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s. Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Mielke, M.M.; Rissman, R.A.; Lista, S.; Vanderstichele, H.; Zetterberg, H.; Lewczuk, P.; Posner, H.; Hall, J.; Johnson, L.; et al. Blood-based biomarkers in Alzheimer disease: Current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimer’s. Dement. 2017, 13, 45–58. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Lanni, C.; Racchi, M.; Memo, M.; Govoni, S.; Uberti, D. p53 at the crossroads between cancer and neurodegeneration. Free Radic. Biol. Med. 2012, 52, 1727–1733. [Google Scholar] [CrossRef]
- Buizza, L.; Cenini, G.; Lanni, C.; Ferrari-Toninelli, G.; Prandelli, C.; Govoni, S.; Buoso, E.; Racchi, M.; Barcikowska, M.; Styczynska, M.; et al. Conformational altered p53 as an early marker of oxidative stress in Alzheimer’s disease. PLoS ONE 2012, 7, e29789. [Google Scholar] [CrossRef] [PubMed]
- Tonello, S.; Stradolini, F.; Abate, G.; Uberti, D.; Serpelloni, M.; Carrara, S.; Sardini, E. Electrochemical detection of different p53 conformations by using nanostructured surfaces. Sci. Rep. 2019, 9, 17347. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, S.; Knights, C.D.; Rao, M.; Yakovlev, A.; Beers, J.; Catania, J.; Avantaggiati, M.L.; Faden, A.I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006, 25, 4084–4096. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell. Biochem. 2014, 85, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef]
- Okuda, Y.; Okuda, M.; Bernard, C.C.A. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2003, 135, 29–37. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Pinkoski, M.J.; Mahboubi, A.; Lin, T.; Han, Z.; Zvaifler, N.J.; Green, D.R.; Firestein, G.S. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am. J. Pathol. 2002, 160, 123–130. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef]
- Jazvinscak Jembrek, M.; Slade, N.; Hof, P.R.; Simic, G. The interactions of p53 with tau and A-beta as potential therapeutic targets for Alzheimer’s disease. Prog. Neurobiol. 2018, 168, 104–127. [Google Scholar] [CrossRef]
- Abate, G.; Frisoni, G.B.; Bourdon, J.-C.; Piccirella, S.; Memo, M.; Uberti, D. The pleiotropic role of p53 in functional/dysfunctional neurons: Focus on pathogenesis and diagnosis of Alzheimer’s disease. Alzheimers. Res. Ther. 2020, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Pati, U. CHIP stabilizes amyloid precursor protein via proteasomal degradation and p53-mediated trans-repression of beta-secretase. Aging Cell 2015, 14, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Checler, F.; Dunys, J.; Pardossi-Piquard, R.; Alves da Costa, C. p53 Is Regulated by and Regulates Members of the γ-Secretase Complex. Neurodegener. Dis. 2010, 7, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Guaita, A.; Colombo, M.; Vaccaro, R.; Fossi, S.; Vitali, S.F.; Forloni, G.; Polito, L.; Davin, A.; Ferretti, V.V.; Villani, S. Brain aging and dementia during the transition from late adulthood to old age: Design and methodology of the “Invece.Ab” population-based study. BMC Geriatr. 2013, 13, 98. [Google Scholar] [CrossRef]
- Galluzzi, S.; Marizzoni, M.; Babiloni, C.; Albani, D.; Antelmi, L.; Bagnoli, C.; Bartres-Faz, D.; Cordone, S.; Didic, M.; Farotti, L.; et al. Clinical and biomarker profiling of prodromal Alzheimer’s disease in workpackage 5 of the Innovative Medicines Initiative PharmaCog project: A ‘European ADNI study. ’ J. Intern. Med. 2016, 279, 576–591. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.J.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s. Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Hort, J.; O’Brien, J.T.; Gainotti, G.; Pirttila, T.; Popescu, B.O.; Rektorova, I.; Sorbi, S.; Scheltens, P. EFNS guidelines for the diagnosis and management of Alzheimer’s disease. Eur. J. Neurol. 2010, 17, 1236–1248. [Google Scholar] [CrossRef]
- Mattsson, N.; Andreasson, U.; Persson, S.; Arai, H.; Batish, S.D.; Bernardini, S.; Bocchio-Chiavetto, L.; Blankenstein, M.A.; Carrillo, M.C.; Chalbot, S.; et al. The Alzheimer’s Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers. Dement. 2011, 7, 386–395.e6. [Google Scholar] [CrossRef]
- McKhann, G.M.; Albert, M.S.; Grossman, M.; Miller, B.; Dickson, D.; Trojanowski, J.Q. Clinical and pathological diagnosis of frontotemporal dementia: Report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch. Neurol. 2001, 58, 1803–1809. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Hansson, O.; Andreasen, N.; Parnetti, L.; Jonsson, M.; Herukka, S.-K.; van der Flier, W.M.; Blankenstein, M.A.; Ewers, M.; et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009, 302, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Chae, W.S.; Kim, H.J.; Shin, H.S.; Kim, S.; Im, J.Y.; Ahn, S.I.; Min, K.D.; Yim, S.J.; Ye, B.S.; et al. Cerebrospinal Fluid Biomarkers for the Diagnosis of Alzheimer Disease in South Korea. Alzheimer Dis. Assoc. Disord. 2017, 31, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Bianchetti, L.; Squazzoni, F.; Casnici, N.; Bianchini, D.; Garrafa, E.; Archetti, C.; Romano, V.; Rozzini, L.; Melchiori, M.; Fiorentini, C.; et al. Social networks and health status in the elderly: The “ANZIANI IN-RETE” population-based study. Aging Clin. Exp. Res. 2017, 29, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.D.; Willett, J.B. Applied Longitudinal Data Analysis: Modeling Change and Event Occurrence; Oxford University Press: New York, NY, USA, 2003; ISBN 9780195152968. [Google Scholar]
- Vezzoli, M.; Ravaggi, A.; Zanotti, L.; Miscioscia, R.A.; Bignotti, E.; Ragnoli, M.; Gambino, A.; Ruggeri, G.; Calza, S.; Sartori, E.; et al. RERT: A Novel Regression Tree Approach to Predict Extrauterine Disease in Endometrial Carcinoma Patients. Sci. Rep. 2017, 7, 10528. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Classification and Regression Trees; CRC press: Boca Raton, FL, USA, 1984. [Google Scholar]
- Vezzoli, M. Exploring the facets of overall job satisfaction through a novel ensemble learning. Electron. J. Appl. Stat. Anal. 2011, 4, 23–38. [Google Scholar]
- Carpita, M.; Vezzoli, M. Statistical evidence of the subjective work quality: The fairness drivers of the job satisfaction. Electron. J. Appl. Stat. Anal. 2012, 5, 89–107. [Google Scholar]
- Savona, R.; Vezzoli, M. Fitting and Forecasting Sovereign Defaults using Multiple Risk Signals. Oxf. Bull. Econ. Stat. 2015, 77, 66–92. [Google Scholar] [CrossRef]
- Arce-Varas, N.; Abate, G.; Prandelli, C.; Martínez, C.; Cuetos, F.; Menéndez, M.; Marziano, M.; Cabrera-García, D.; Fernández-Sánchez, M.T.; Novelli, A.; et al. Comparison of extracellular and intracellular blood compartments highlights redox alterations in Alzheimer’s and mild cognitive impairment patients. Curr. Alzheimer Res. 2017, 14, 112–122. [Google Scholar] [CrossRef]
- Dancelli, L.; Manisera, M.; Vezzoli, M. On two classes of Weighted Rank Correlation measures deriving from the Spearman’s ρ. In Statistical Models for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2013; pp. 107–114. [Google Scholar]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35, 7475–7484. [Google Scholar] [CrossRef]
- Feng, Z.; Lin, M.; Wu, R. The Regulation of Aging and Longevity: A New and Complex Role of p53. Genes Cancer 2011, 2, 443–452. [Google Scholar] [CrossRef]
- Uberti, D.; Carsana, T.; Bernardi, E.; Rodella, L.; Grigolato, P.; Lanni, C.; Racchi, M.; Govoni, S.; Memo, M. Selective impairment of p53-mediated cell death in fibroblasts from sporadic Alzheimer’s disease patients. J. Cell Sci. 2002, 115, 3131–3138. [Google Scholar] [PubMed]
- Lanni, C.; Racchi, M.; Mazzini, G.; Ranzenigo, A.; Polotti, R.; Sinforiani, E.; Olivari, L.; Barcikowska, M.; Styczynska, M.; Kuznicki, J.; et al. Conformationally altered p53: A novel Alzheimer’s disease marker? Mol. Psychiatry 2008, 13, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Lanni, C.; Racchi, M.; Stanga, S.; Mazzini, G.; Ranzenigo, A.; Polotti, R.; Memo, M.; Govoni, S.; Uberti, D. Unfolded p53 in blood as a predictive signature signature of the transition from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers. Dis. 2010, 20, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Farlow, M.; Aisen, P.S. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 1460. [Google Scholar] [CrossRef] [PubMed]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.K.; Smith, A.M.; Walsh, J.R.; Simon, A.J.; Edgar, C.; Jack, C.R.; Holtzman, D.; Russell, D.; Hill, D.; Grosset, D.; et al. Machine learning for comprehensive forecasting of Alzheimer’s Disease progression. Sci. Rep. 2019, 9, 13622. [Google Scholar] [CrossRef]
- Lanni, C.; Nardinocchi, L.; Puca, R.; Stanga, S.; Uberti, D.; Memo, M.; Govoni, S.; D’Orazi, G.; Racchi, M. Homeodomain interacting protein kinase 2: A target for Alzheimer’s beta amyloid leading to misfolded p53 and inappropriate cell survival. PLoS ONE 2010, 5, e10171. [Google Scholar] [CrossRef]
- Meplan, C.; Richard, M.J.; Hainaut, P. Redox signalling and transition metals in the control of the p53 pathway. Biochem. Pharmacol. 2000, 59, 25–33. [Google Scholar] [CrossRef]
- Tonello, S.; Serpelloni, M.; Lopomo, N.F.; Sardini, E.; Abate, G.; Uberti, D.L. Preliminary study of a low-cost point-of-care testing system using screen-printed biosensors: For early biomarkers detection related to Alzheimer Disease. In Proceedings of the 2016 IEEE International Symposium on Medical Measurements and Applications, MeMeA 2016, Benevento, Italy, 15–18 May 2016. [Google Scholar]
- Lowe, J.; Shatz, M.; Resnick, M.A.; Menendez, D. Modulation of immune responses by the tumor suppressor p53. Biodiscovery 2013, 8, e8947. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, J.; Feng, Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013, 3, 9. [Google Scholar] [CrossRef]
- Abate, G.; Vezzoli, M.; Sandri, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Mitochondria and cellular redox state on the route from ageing to Alzheimer’s disease. Mech. Ageing Dev. 2020, 192, 111385. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| InveCe.Ab | PharmaCog/E-ADNI | ||
|---|---|---|---|
| No. plasma samples | 264 * | 111 | |
| Classification | CN | MCI | aMCI |
| N. subjects | 64 | 26 | 111 a |
| Sex: female, n (%) | 37 (57.81%) | 8(30.77%) | 64 (57.66%) |
| Age, mean (SD) | 73 (1.22) | 73.28 (1.36) | 69.12 (7.55) |
| APOEε4 status, n (%) | 14 (21.87%) | 8 (30.77%) | 52 (46.85%) |
| Level of education (SD) b | 1.14 (0.65) | 1.16 (0.54) | 1.37 (0.50) |
| MMSE (SD) | 27.56 (2.28) | 26.5 (2.11) | 26.41 (1.83) |
| Conversion to AD, N | 10/64 | 9/26 | 18/111 |
| Conversion time (min–max), months | 24–48 | 24–48 | 6–30 |
| AD-related biomarkers | |||
| CSF Aβ42 level (SD), pg/mL | 676.41 (290.72) | ||
| CSF p-Tau level (SD), pg/mL | 70.31 (36.98) | ||
| CSF t-Tau level (SD), pg/mL | 503.82 (373.44) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abate, G.; Vezzoli, M.; Polito, L.; Guaita, A.; Albani, D.; Marizzoni, M.; Garrafa, E.; Marengoni, A.; Forloni, G.; Frisoni, G.B.; et al. A Conformation Variant of p53 Combined with Machine Learning Identifies Alzheimer Disease in Preclinical and Prodromal Stages. J. Pers. Med. 2021, 11, 14. https://doi.org/10.3390/jpm11010014
Abate G, Vezzoli M, Polito L, Guaita A, Albani D, Marizzoni M, Garrafa E, Marengoni A, Forloni G, Frisoni GB, et al. A Conformation Variant of p53 Combined with Machine Learning Identifies Alzheimer Disease in Preclinical and Prodromal Stages. Journal of Personalized Medicine. 2021; 11(1):14. https://doi.org/10.3390/jpm11010014
Chicago/Turabian StyleAbate, Giulia, Marika Vezzoli, Letizia Polito, Antonio Guaita, Diego Albani, Moira Marizzoni, Emirena Garrafa, Alessandra Marengoni, Gianluigi Forloni, Giovanni B. Frisoni, and et al. 2021. "A Conformation Variant of p53 Combined with Machine Learning Identifies Alzheimer Disease in Preclinical and Prodromal Stages" Journal of Personalized Medicine 11, no. 1: 14. https://doi.org/10.3390/jpm11010014
APA StyleAbate, G., Vezzoli, M., Polito, L., Guaita, A., Albani, D., Marizzoni, M., Garrafa, E., Marengoni, A., Forloni, G., Frisoni, G. B., Cummings, J. L., Memo, M., & Uberti, D. (2021). A Conformation Variant of p53 Combined with Machine Learning Identifies Alzheimer Disease in Preclinical and Prodromal Stages. Journal of Personalized Medicine, 11(1), 14. https://doi.org/10.3390/jpm11010014