The Genetics of Spondyloarthritis

 ,
,  ,
, 
Abstract
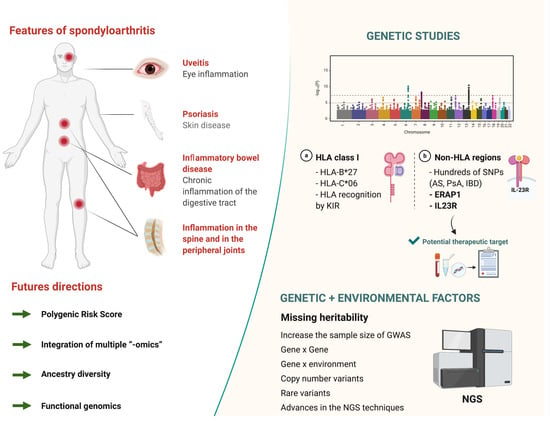
1. Introduction
2. Genetic Epidemiology of SpA
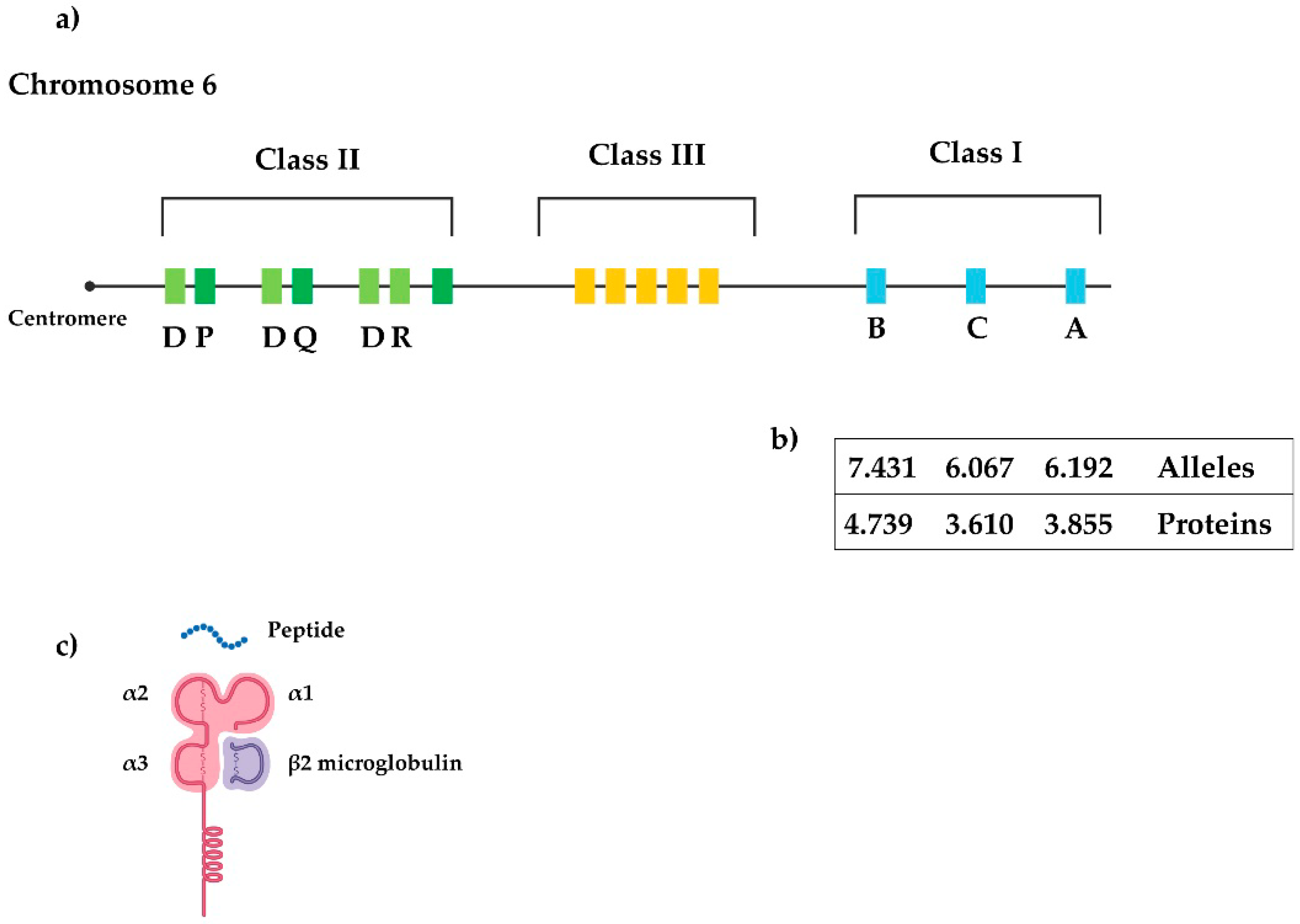
3. Major Histocompatibility Complex
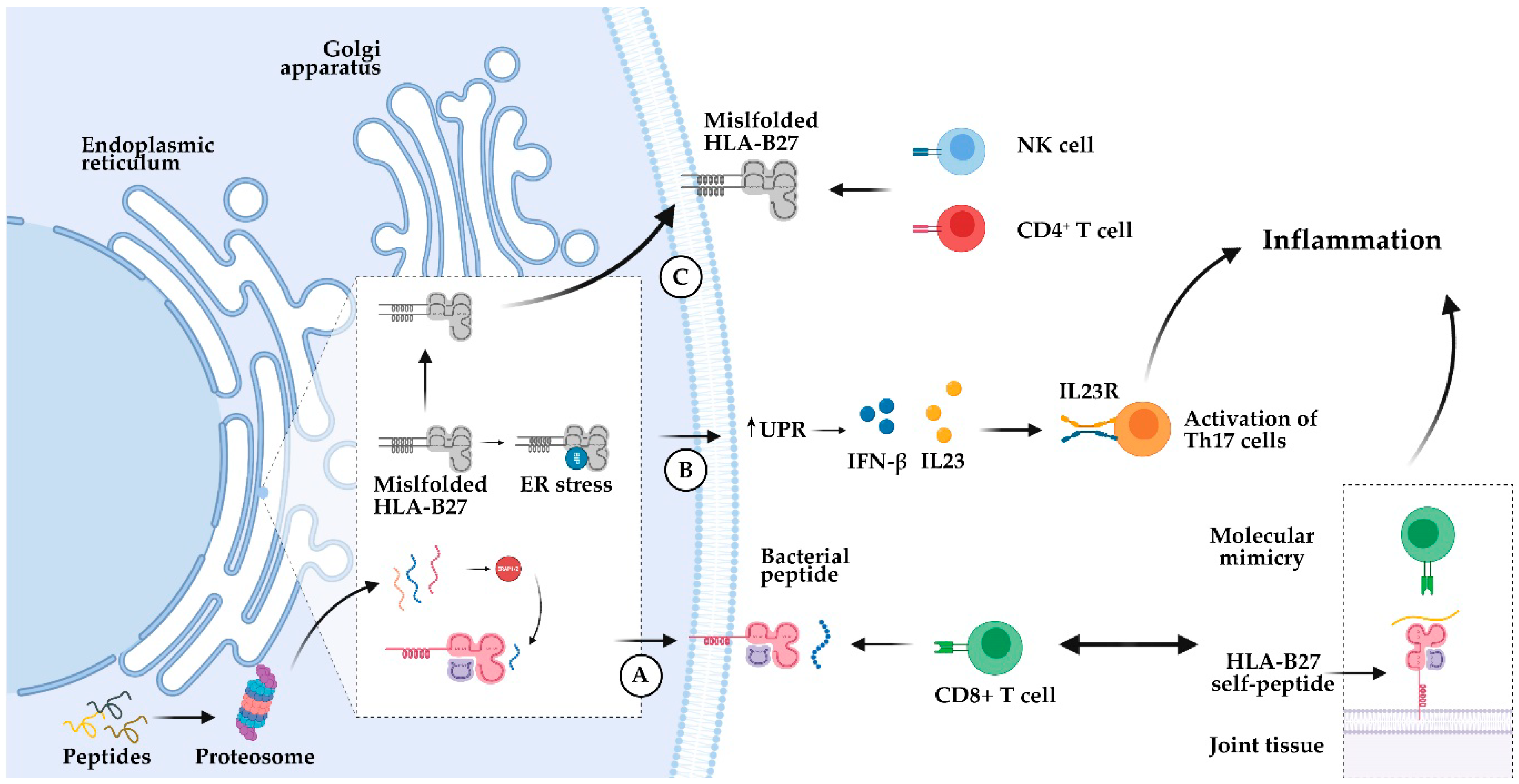
3.1. HLA-B27
3.2. Other HLA Class I Genes
3.3. Recognition by NK Cell Receptors
3.4. MHC Class II Region
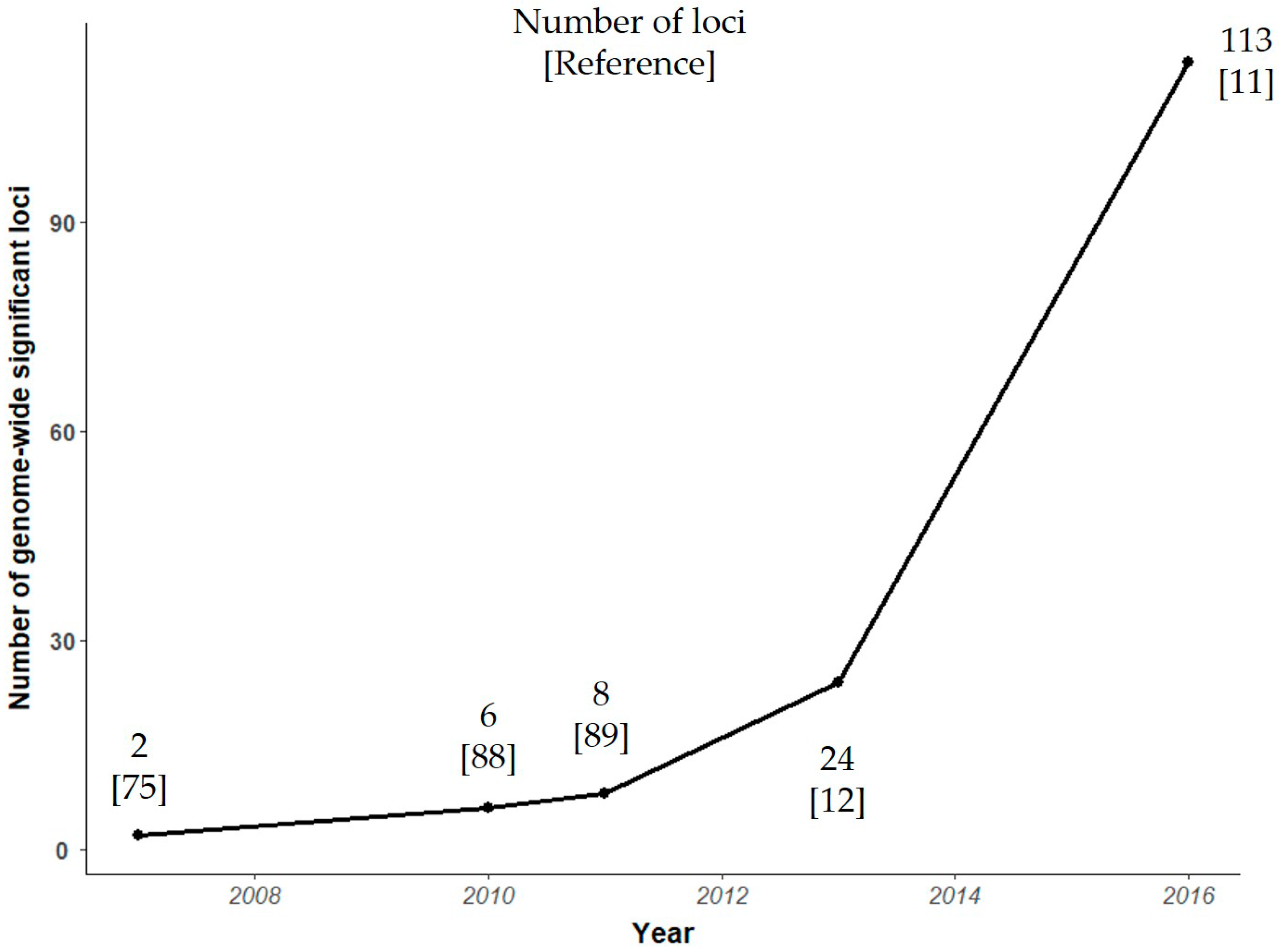
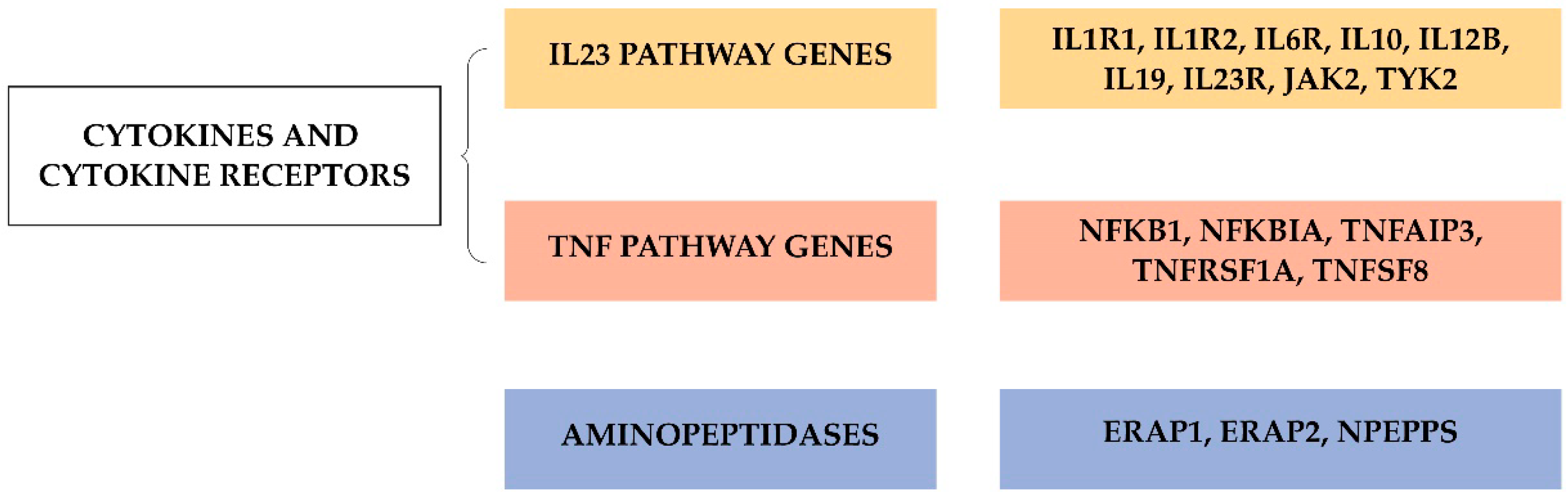
4. Genome Wide Association Studies
4.1. Current Status of Variant Discovery
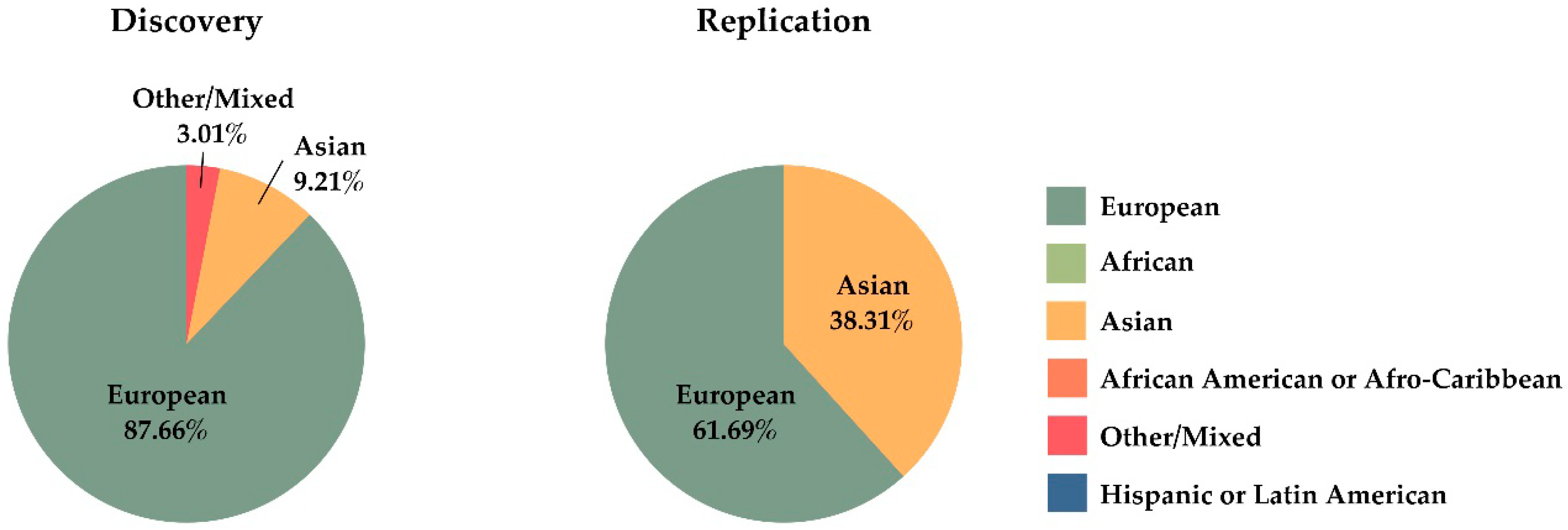
4.2. Missing Heritability
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sparks, J.A.; Costenbader, K.H. Genetics, environment, and gene-environment interactions in the development of systemic rheumatic diseases. Rheum. Dis. Clin. N. Am. 2014, 40, 637–657. [Google Scholar] [CrossRef] [PubMed]
- Schlosstein, L.; Terasaki, P.I.; Bluestone, R.; Pearson, C.M. High Association of an HL-A Antigen, W27, with Ankylosing Spondylitis. N. Engl. J. Med. 1973, 288, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M.F.P.; James, D.C.O. Human lymphocyte antigen association in ankylosing spondylitis. Nature 1973, 242, 121. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Brown, M.A. Progress of genome-wide association studies of ankylosing spondylitis. Clin. Transl. Immunol. 2017, 6, e163. [Google Scholar] [CrossRef]
- De Blecourt, J.J.; Polman, A.; de Blécourt-Meindersma, T. Hereditary factors in rheumatoid arthritis and ankylosing spondylitis. Ann. Rheum. Dis. 1961, 20, 215–220. [Google Scholar] [CrossRef]
- Brown, M.A.; Laval, S.H.; Brophy, S.; Calin, A. Recurrence risk modelling of the genetic susceptibility to ankylosing spondylitis. Ann. Rheum. Dis. 2000, 59, 883–886. [Google Scholar] [CrossRef]
- Dernis, E.; Said-Nahal, R.; D’agostino, M.A.; Aegerter, P.; Dougados, M.; Breban, M. Recurrence of spondylarthropathy among firstdegree relatives of patients: A systematic crosssectional Study. Ann. Rheum. Dis. 2009, 68, 502–507. [Google Scholar] [CrossRef]
- Brown, M.A.; Kennedy, L.G.; MacGregor, A.J.; Darke, C.; Duncan, E.; Shatford, J.L.; Taylor, A.; Calin, A.; Wordsworth, P. Susceptibility to ankylosing spondylitis in twins: The role of genes, HLA, and the environment. Arthritis Rheum. 1997, 40, 1823–1828. [Google Scholar] [CrossRef]
- Thomas, G.P.; Brown, M.A. Genetics and genomics of ankylosing spondylitis. Immunol. Rev. 2010, 233, 162–180. [Google Scholar] [CrossRef]
- Dean, L.E.; Jones, G.T.; Macdonald, A.G.; Downham, C.; Sturrock, R.D.; Macfarlane, G.J. Global prevalence of ankylosing spondylitis. Rheumatology 2014, 53, 650–657. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Jostins, L.; Spain, S.L.; Cortes, A.; Bethune, J.; Han, B.; Park, Y.R.; Raychaudhuri, S.; Pouget, J.G.; Hübenthal, M.; et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 2016, 48, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Hadler, J.; Pointon, J.P.; Robinson, P.C.; Karaderi, T.; Leo, P.; Cremin, K.; Pryce, K.; Harris, J.; Lee, S.; et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 2013, 45, 730–738. [Google Scholar] [PubMed]
- McGonagle, D.; Aydin, S.Z.; Gül, A.; Mahr, A.; Direskeneli, H. ’MHC-I-opathy’—Unified concept for spondyloarthritis and Behçet disease. Nat. Rev. Rheumatol. 2015, 11, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.; Kay, L.J.; Lynch, S.A.; Walker, D.J. Recurrence risk for psoriasis and psoriatic arthritis within sibships. Rheumatology 2005, 44, 773–776. [Google Scholar] [CrossRef]
- Chandran, V.; Schentag, C.T.; Brockbank, J.E.; Pellett, F.J.; Shanmugarajah, S.; Toloza, S.M.A.; Rahman, P.; Gladman, D.D. Familial aggregation of psoriatic arthritis. Ann. Rheum. Dis. 2009, 68, 664–667. [Google Scholar] [CrossRef]
- Winchester, R.; Minevich, G.; Steshenko, V.; Kirby, B.; Kane, D.; Greenberg, D.A.; FitzGerald, O. HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum. 2012, 64, 1134–1144. [Google Scholar] [CrossRef]
- Stuart, P.E.; Nair, R.P.; ELinghaus, E.; Ding, J.; Tejasvi, T.; GudjonSon, J.E.; Li, Y.; Weidinger, S.; Eberlein, B.; Gieger, C.; et al. Genome-wide asociation analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010, 42, 1000–1004. [Google Scholar] [CrossRef]
- Stuart, P.E.; Nair, R.P.; Tsoi, L.C.; Tejasvi, T.; Das, S.; Kang, H.M.; Ellinghaus, E.; Chandran, V.; Callis-Duffin, K.; Ike, R.; et al. Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. Am. J. Hum. Genet. 2015, 97, 816–836. [Google Scholar] [CrossRef]
- Halme, L.; Paavola-Sakki, P.; Turunen, U.; Lappalainen, M.; Färkkilä, M.; Kontula, K. Family and twin studies in inflammatory bowel disease. World J. Gastroenterol. 2006, 12, 3668–3672. [Google Scholar] [CrossRef]
- Annese, V. Genetics and epigenetics of IBD. Pharmacol. Res. 2020, 159, 104892. [Google Scholar] [CrossRef]
- Trowsdale, J.; Knight, J.C. Major histocompatibility complex genomics and human disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Guethlein, L.A.; Cereb, N.; Yang, S.Y.; Norman, P.J.; Marsh, S.G.E.; Parham, P. Distinguishing functional polymorphism from random variation in the sequences of >10,000 HLA-A, -B and -C alleles. PLoS Genet. 2017, 13, e1006862. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Roces, S.; Alvarez, M.V.; Gonzalez, S.; Dieye, A.; Makni, H.; Woodfield, D.G.; Housan, L.; Konenkov, V.; Abbadi, M.C.; Grunnet, N.; et al. HLA-B27 polymorphism and worldwide susceptibility to ankylosing spondylitis. Tissue Antigens 1997, 49, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A. Remarkable polymorphism of HLA-B27: An ongoing saga. Curr. Rheumatol. Rep. 2010, 12, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Reveille, J.D.; Maganti, R.M. Subtypes of HLA-B27: History and implications in the pathogenesis of ankylosing spondylitis. Adv. Exp. Med. Biol. 2009, 649, 159–176. [Google Scholar] [PubMed]
- Colbert, R.A.; Navid, F.; Gill, T. The role of HLA-B*27 in spondyloarthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 797–815. [Google Scholar] [CrossRef]
- López-Larrea, C.; Sujirachato, K.; Mehra, N.K.; Chiewsilp, P.; Isarangkura, D.; Kanga, U.; Dominguez, O.; Coto, E.; Penã, M.; Setién, F.; et al. HLA-B27 subtypes in Asian patients with ankylosing spondylitis Evidence for new associations. Tissue Antigens 1995, 45, 169–176. [Google Scholar] [CrossRef]
- Paladini, F.; Taccari, E.; Fiorillo, M.T.; Cauli, A.; Passiu, G.; Mathieu, A.; Punzi, L.; Lapadula, G.; Scarpa, R.; Sorrentino, R. Distribution of HLA-B27 subtypes in Sardinia and continental Italy and their association with spondylarthropathies. Arthritis Rheum. 2005, 52, 3319–3321. [Google Scholar] [CrossRef]
- Benjamin, R.; Parham, P. Guilt by association: HLA-B27 and ankylosing spondylitis. Immunol. Today 1990, 11, 137–142. [Google Scholar] [CrossRef]
- Kollnberger, S.; Bowness, P. The role of B27 heavy chain dimer immune receptor interactions in spondyloarthritis. Adv. Exp. Med. Biol. 2009, 649, 277–285. [Google Scholar]
- Bowness, P.; Ridley, A.; Shaw, J.; Chan, A.T.; Wong-Baeza, I.; Fleming, M.; Cummings, F.; McMichael, A.; Kollnberger, S. Th17 Cells Expressing KIR3DL2+ and Responsive to HLA-B27 Homodimers Are Increased in Ankylosing Spondylitis. J. Immunol. 2011, 186, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Peña, R.; López-Vázquez, A.; López-Larrea, C. Old and new HLA associations with ankylosing spondylitis. Tissue Antigens 2012, 80, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, R.B.; Sian, T.C.C.L.K.; Wilmann, P.G.; Dudek, N.L.; Purcell, A.W. Revisiting the arthritogenic peptide theory: Quantitative not qualitative changes in the peptide repertoire of HLA-B27 allotypes. Arthritis Rheumatol. 2015, 67, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Colbert, R.A.; DeLay, M.L.; Layh-Schmitt, G.; Sowders, D.P. HLA-B27 misfolding and spondyloarthropathies. Adv. Exp. Med. Biol. 2009, 649, 217–234. [Google Scholar]
- Costello, M.E.; Ciccia, F.; Willner, D.; Warrington, N.; Robinson, P.C.; Gardiner, B.; Marshall, M.; Kenna, T.J.; Triolo, G.; Brown, M.A. Brief Report: Intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol. 2015, 67, 686–691. [Google Scholar] [CrossRef]
- Breban, M.; Tap, J.; Leboime, A.; Said-Nahal, R.; Langella, P.; Chiocchia, G.; Furet, J.P.; Sokol, H. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 1614–1622. [Google Scholar] [CrossRef]
- Breban, M.; Beaufrère, M.; Glatigny, S. The microbiome in spondyloarthritis. Best Pract. Res. Clin. Rheumatol. 2019, 33, 101495. [Google Scholar] [CrossRef]
- Asquith, M.J.; Stauffer, P.; Davin, S.; Mitchell, C.; Lin, P.; Rosenbaum, J.T. Perturbed Mucosal Immunity and Dysbiosis Accompany Clinical Disease in a Rat Model of Spondyloarthritis. Arthritis Rheumatol. 2016, 68, 2151–2162. [Google Scholar] [CrossRef]
- Ciccia, F.; Bombardieri, M.; Principato, A.; Giardina, A.; Tripodo, C.; Porcasi, R.; Peralta, S.; Franco, V.; Giardina, E.; Craxi, A.; et al. Overexpression of interleukin-23, but Not interleukin-17, as an immunologic signature of Subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009, 60, 955–965. [Google Scholar] [CrossRef]
- Cortes, A.; Pulit, S.L.; Leo, P.J.; Pointon, J.J.; Robinson, P.C.; Weisman, M.H.; Ward, M.; Gensler, L.S.; Zhou, X.; Garchon, H.-J.; et al. Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat. Commun. 2015, 6, 7146. [Google Scholar] [CrossRef]
- Reveille, J.D.; Zhou, X.; Lee, M.J.; Weisman, M.H.; Yi, L.; Gensler, L.S.; Zou, H.; Ward, M.M.; Ishimori, M.L.; Learch, T.J.; et al. HLA class i and II alleles in susceptibility to ankylosing spondylitis. Ann. Rheum. Dis. 2019, 78, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Bang, S.Y.; Lee, S.; Lee, H.S.; Shim, S.C.; Kang, Y.M.; Suh, C.H.; Sun, C.; Nath, S.K.; Bae, S.C.; et al. An HLA-C amino-acid variant in addition to HLA-B*27 confers risk for ankylosing spondylitis in the Korean population. Arthritis Res. Ther. 2015, 17, 342. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Jepson, A.; Young, A.; Whittle, H.C.; Greenwood, B.M.; Wordsworth, B.P. Ankylosing spondylitis in West Africans—Evidence for a non-HLA-B27 protective effect. Ann. Rheum. Dis. 1997, 56, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Reveille, J.D.; Witter, J.P.; Weisman, M.H. Prevalence of axial spondylarthritis in the United States: Estimates from a cross-sectional Survey. Arthritis Care Res. 2012, 64, 905–910. [Google Scholar] [CrossRef]
- Díaz-Peña, R.; Blanco-Gelaz, M.A.; Njobvu, P.; López-Vazquez, A.; Suárez-Álvarez, B.; López-Larrea, C. Influence of HLA-B*5703 and HLA-B*1403 on susceptibility to spondyloarthropathies in the Zambian population. J. Rheumatol. 2008, 35, 2236–2240. [Google Scholar] [CrossRef]
- Díaz-Peña, R.; Ouédraogo, D.D.; López-Vázquez, A.; Sawadogo, S.A.; López-Larrea, C. Ankylosing spondylitis in three Sub-Saharan populations: HLA-B*27 and HLA-B*14 contribution. Tissue Antigens 2012, 80, 14–15. [Google Scholar] [CrossRef]
- Benegas, M.; Muñoz-Gomariz, E.; Font, P.; Burgos-Vargas, R.; Chaves, J.; Palleiro, D.; Maldonado Cocco, J.; Gutiérrez, M.; Sáenz, R.; Steckmen, I.; et al. Comparison of the clinical expression of patients with ankylosing spondylitis from Europe and Latin America. J. Rheumatol. 2012, 39, 2315–2320. [Google Scholar] [CrossRef]
- Hsu, K.C.; Chida, S.; Geraghty, D.E.; Dupont, B. The killer cell immunoglobulin-like receptor (KIR) genomic region: Gene-order, haplotypes and allelic polymorphism. Immunol. Rev. 2002, 190, 40–52. [Google Scholar] [CrossRef]
- Cella, M.; Longo, A.; Ferrara, G.B.; Strominger, J.L.; Colonna, M. NK3-specific natural killer cells are selectively inhibited by Bw4-positive HLA alleles with isoleucine 80. J. Exp. Med. 1994, 180, 1235–1242. [Google Scholar] [CrossRef]
- Colonna, M.; Samaridis, J.; Cella, M.; Allen, R.L.; Callaghan, C.A.O.; Dunbar, R.; Ogg, G.S.; Cerundolo, V.; Rolink, A.; Angman, L. Cutting Edge: Human Myelomonocytic Cells Express an Inhibitory Receptor for Classical and Nonclassical MHC Class I Molecules. J. Immunol. 1998, 160, 3096–3100. [Google Scholar]
- Kollnberger, S.; Bird, L.; Sun, M.Y.; Retiere, C.; Braud, V.M.; McMichael, A.; Bowness, P. Cell-surface expression and immune receptor recognition of HLA-B27 homodimers. Arthritis Rheum. 2002, 46, 2972–2982. [Google Scholar] [CrossRef] [PubMed]
- Wong-Baeza, I.; Ridley, A.; Shaw, J.; Hatano, H.; Rysnik, O.; McHugh, K.; Piper, C.; Brackenbridge, S.; Fernandes, R.; Chan, A.; et al. KIR3DL2 Binds to HLA-B27 Dimers and Free H Chains More Strongly than Other HLA Class I and Promotes the Expansion of T Cells in Ankylosing Spondylitis. J. Immunol. 2013, 190, 3216–3224. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Nelson, G.; Lee, J.-H.; Pellett, F.; Gao, X.; Wade, J.; Wilson, M.J.; Trowsdale, J.; Gladman, D.; Carrington, M. Cutting Edge: Susceptibility to Psoriatic Arthritis: Influence of Activating Killer Ig-Like Receptor Genes in the Absence of Specific HLA-C Alleles. J. Immunol. 2002, 169, 2818–2822. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.; Meenagh, A.; Sleator, C.; Cook, D.; Fernandez-Vina, M.; Bowcock, A.M.; Middleton, D. Activating killer cell immunoglobulin-like receptor gene KIR2DS1 is associated with psoriatic arthritis. Hum. Immunol. 2005, 66, 836–841. [Google Scholar] [CrossRef]
- Holm, S.J.; Sakuraba, K.; Mallbris, L.; Wolk, K.; Ståhle, M.; Sánchez, F.O. Distinct HLA-C/KIR genotype profile associates with guttate psoriasis. J. Investig. Dermatol. 2005, 125, 721–730. [Google Scholar] [CrossRef]
- Lopez-Larrea, C.; Blanco-Gelaz, M.A.; Torre-Alonso, J.C.; Armas, J.B.; Suarez-Alvarez, B.; Pruneda, L.; Couto, A.R.; Gonzalez, S.; Lopez-Vázquez, A.; Martinez-Borra, J. Contribution of KIR3DL1/3DS1 to ankylosing spondylitis in human leukocyte antigen-B27 Caucasian populations. Arthritis Res. Ther. 2006, 8, R101. [Google Scholar] [CrossRef]
- Díaz-Peña, R.; Blanco-Gelaz, M.A.; Suárez-Álvarez, B.; Martínez-Borra, J.; López-Vázquez, A.; Alonso-Arias, R.; Brüges-Armas, J.; Vidal-Castiñeira, J.R.; López-Larrea, C. Activating KIR genes are associated with ankylosing spondylitis in Asian populations. Hum. Immunol. 2008, 69, 437–442. [Google Scholar] [CrossRef]
- Harvey, D.; Pointon, J.J.; Sleator, C.; Meenagh, A.; Farrar, C.; Sun, J.Y.; Senitzer, D.; Middleton, D.; Brown, M.A.; Wordsworth, B.P. Analysis of killer immunoglobulin-like receptor genes in ankylosing spondylitis. Ann. Rheum. Dis. 2009, 68, 595–598. [Google Scholar] [CrossRef]
- Zvyagin, I.V.; Mamedov, I.Z.; Britanova, O.V.; Staroverov, D.B.; Nasonov, E.L.; Bochkova, A.G.; Chkalina, A.V.; Kotlobay, A.A.; Korostin, D.O.; Rebrikov, D.V.; et al. Contribution of functional KIR3DL1 to ankylosing spondylitis. Cell. Mol. Immunol. 2010, 7, 471–476. [Google Scholar] [CrossRef]
- Jiao, Y.L.; Zhang, B.C.; You, L.; Li, J.F.; Zhang, J.; Ma, C.Y.; Cui, B.; Wang, L.C.; Chen, Z.J.; Zhao, Y.R. Polymorphisms of KIR gene and HLA-C alleles: Possible association with susceptibility to HLA-B27-positive patients with ankylosing spondylitis. J. Clin. Immunol. 2010, 30, 840–844. [Google Scholar] [CrossRef]
- Díaz-Peña, R.; Vidal-Castiñeira, J.R.; Alonso-Arias, R.; Suarez-Alvarez, B.; Vicario, J.L.; Solana, R.; Collantes, E.; López-Vázquez, A.; Martínez-Borra, J.; López-Larrea, C. Association of the KIR3DS1*013 and KIR3DL1*004 alleles with susceptibility to ankylosing Spondylitis. Arthritis Rheum. 2010, 62, 1000–1006. [Google Scholar] [CrossRef]
- Tajik, N.; Shahsavar, F.; Poormoghim, H.; Radjabzadeh, M.F.; Mousavi, T.; Jalali, A. KIR3DL1+HLA-B Bw4 Ile80 and KIR2DS1+HLA-C2 combinations are both associated with ankylosing spondylitis in the Iranian population. Int. J. Immunogenet. 2011, 38, 403–409. [Google Scholar] [CrossRef]
- Wang, S.; Li, G.; Ge, R.; Duan, Z.; Zeng, Z.; Zhang, T.; Gao, J.; Yang, T.; Liu, S.; Wu, S.; et al. Association of KIR genotype with susceptibility to HLA-B27-positive ankylosing spondylitis. Mod. Rheumatol. 2013, 23, 538–541. [Google Scholar] [CrossRef]
- Moon, S.J.; Oh, E.J.; Kim, Y.; Kim, K.S.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Kim, H.Y.; Park, S.H. Diversity of killer cell immunoglobulin-like receptor genes in uveitis associated with autoimmune diseases: Ankylosing spondylitis and behçet disease. Ocul. Immunol. Inflamm. 2013, 21, 135–143. [Google Scholar] [CrossRef]
- Chandran, V.; Bull, S.B.; Pellett, F.J.; Ayearst, R.; Pollock, R.A.; Gladman, D.D. Killer-cell immunoglobulin-like receptor gene polymorphisms and susceptibility to psoriatic arthritis. Rheumatology 2014, 53, 233–239. [Google Scholar] [CrossRef]
- Díaz-Peña, R.; Vidal-Castiñeira, J.R.; Mulero, J.; Sánchez, A.; Queiro, R.; López-Larrea, C. Activating killer immunoglobulin-like receptors genes are associated with increased susceptibility to ankylosing spondylitis. Clin. Exp. Immunol. 2015, 180, 201–206. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Jamshidi, A.R.; Karami, J.; Mohseni, A.; Amirzargar, A.A.; Farhadi, E.; Ahmadzadeh, N.; Nicknam, M.H. Analysis of killer cell immunoglobulin-like receptor genes and their HLA ligands in Iranian patients with Ankylosing Spondylitis. Iran. J. Allergy Asthma Immunol. 2016, 15, 27–38. [Google Scholar]
- Berinstein, J.; Pollock, R.; Pellett, F.; Thavaneswaran, A.; Chandran, V.; Gladman, D.D. Association of variably expressed KIR3dl1 alleles with psoriatic disease. Clin. Rheumatol. 2017, 36, 2261–2266. [Google Scholar] [CrossRef]
- Wang, C.M.; Wang, S.H.; Wu, Y.J.; Lin, J.C.; Wu, J.; Chen, J.Y. Human leukocyte antigen c*12:02:02 and killer immunoglobulin-like receptor 2dl5 are distinctly associated with ankylosing spondylitis in the Taiwanese. Int. J. Mol. Sci. 2017, 18, 1775. [Google Scholar] [CrossRef]
- Sun, H.S.; Liu, D.X.; Bai, Y.Y.; Hu, N.W. Disease-association of different killer cell immunoglobulin-like receptors (KIR) and HLA-C gene combinations in reactive arthritis. Mod. Rheumatol. 2019, 29, 531–537. [Google Scholar] [CrossRef]
- Enciso-Vargas, M.; Alvarado-Ruíz, L.; Suárez-Villanueva, A.S.; Macías-Barragán, J.; Montoya-Buelna, M.; Oceguera-Contreras, E.; Alvarado-Navarro, A.; Graciano-Machuca, O. Association Study between Psoriatic Arthritis and Killer Immunoglobulin-Like Receptor (KIR) Genes: A Meta-Analysis. Immunol. Investig. 2020, 1–12. [Google Scholar] [CrossRef]
- Díaz-Peña, R.; Aransay, A.M.; Bruges-Armas, J.; López-Vázquez, A.; Rodríguez-Ezpeleta, N.; Mendibil, I.; Sánchez, A.; Torre-Alonso, J.C.; Bettencourt, B.F.; Mulero, J.; et al. Fine mapping of a major histocompatibility complex in ankylosing spondylitis: Association of the HLA-DPA1 and HLA-DPB1 regions. Arthritis Rheum. 2011, 63, 3305–3312. [Google Scholar] [CrossRef]
- Díaz-Peña, R.; Castro-Santos, P.; Aransay, A.M.; Brüges-Armas, J.; Pimentel-Santos, F.M.; López-Larrea, C. Genetic study confirms association of HLA-DPA1*01:03 subtype with ankylosing spondylitis in HLA-B27-positive populations. Hum. Immunol. 2013, 74, 764–767. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007, 39, 1329–1337. [Google Scholar]
- Costantino, F.; Breban, M.; Garchon, H.J. Genetics and Functional Genomics of Spondyloarthritis. Front. Immunol. 2018, 9, 2933. [Google Scholar] [CrossRef]
- Chang, S.C.; Momburg, F.; Bhutani, N.; Goldberg, A.L. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 17107–17112. [Google Scholar] [CrossRef]
- Martín-Esteban, A.; Sanz-Bravo, A.; Guasp, P.; Barnea, E.; Admon, A.; López de Castro, J.A. Separate effects of the ankylosing spondylitis associated ERAP1 and ERAP2 aminopeptidases determine the influence of their combined phenotype on the HLA-B*27 peptidome. J. Autoimmun. 2017, 79, 28–38. [Google Scholar] [CrossRef]
- Hanson, A.L.; Cuddihy, T.; Haynes, K.; Loo, D.; Morton, C.J.; Oppermann, U.; Leo, P.; Thomas, G.P.; Lê Cao, K.A.; Kenna, T.J.; et al. Genetic Variants in ERAP1 and ERAP2 Associated With Immune-Mediated Diseases Influence Protein Expression and the Isoform Profile. Arthritis Rheumatol. 2018, 70, 255–265. [Google Scholar] [CrossRef]
- Maben, Z.; Arya, R.; Rane, D.; An, W.F.; Metkar, S.; Hickey, M.; Bender, S.; Ali, A.; Nguyen, T.T.; Evnouchidou, I.; et al. Discovery of Selective Inhibitors of Endoplasmic Reticulum Aminopeptidase 1. J. Med. Chem. 2020, 63, 103–121. [Google Scholar] [CrossRef]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef]
- Cargill, M.; Schrodi, S.J.; Chang, M.; Garcia, V.E.; Brandon, R.; Callis, K.P.; Matsunami, N.; Ardlie, K.G.; Civello, D.; Catanese, J.J.; et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 2007, 80, 273–290. [Google Scholar] [CrossRef]
- Baeten, D.; Baraliakos, X.; Braun, J.; Sieper, J.; Emery, P.; Van Der Heijde, D.; McInnes, I.; Van Laar, J.M.; Landewé, R.; Wordsworth, P.; et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: A randomised, double-blind, placebo-controlled trial. Lancet 2013, 382, 1705–1713. [Google Scholar] [CrossRef]
- Vecellio, M.; Roberts, A.R.; Cohen, C.J.; Cortes, A.; Knight, J.C.; Bowness, P.; Wordsworth, B.P. The genetic association of RUNX3 with ankylosing spondylitis can be explained by allele-specific effects on IRF4 recruitment that alter gene expression. Ann. Rheum. Dis. 2016, 75, 1534–1540. [Google Scholar] [CrossRef]
- Apel, M.; Uebe, S.; Bowes, J.; Giardina, E.; Korendowych, E.; Juneblad, K.; Pasutto, F.; Ekici, A.B.; McManus, R.; Ho, P.; et al. Variants in RUNX3 Contribute to Susceptibility to Psoriatic Arthritis, Exhibiting Further Common Ground with Ankylosing Spondylitis. Arthritis Rheum. 2013, 65, 1224–1231. [Google Scholar] [CrossRef]
- Cruz-Guilloty, F.; Pipkin, M.E.; Djuretic, I.M.; Levanon, D.; Lotem, J.; Lichtenheld, M.G.; Groner, Y.; Rao, A. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 2009, 206, 51–59. [Google Scholar] [CrossRef]
- Lotem, J.; Levanon, D.; Negreanu, V.; Bauer, O.; Hantisteanu, S.; Dicken, J.; Groner, Y. Runx3 at the interface of immunity, inflammation and cancer. Biochim. Biophys. Acta-Rev. Cancer 2015, 1855, 131–143. [Google Scholar] [CrossRef]
- Reveille, J.D.; Sims, A.M.; Danoy, P.; Evans, D.M.; Leo, P.; Pointon, J.J.; Jin, R.; Zhou, X.; Bradbury, L.A.; Appleton, L.H.; et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat. Genet. 2010, 42, 123–127. [Google Scholar]
- Evans, D.M.; Spencer, C.C.; Pointon, J.J.; Su, Z.; Harvey, D.; Kochan, G.; Oppermann, U.; Dilthey, A.; Pirinen, M.; Stone, M.; et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat. Genet. 2011, 43, 761–767. [Google Scholar] [CrossRef]
- Liu, Y.; Helms, C.; Liao, W.; Zaba, L.C.; Duan, S.; Gardner, J.; Wise, C.; Miner, A.; Malloy, M.J.; Pullinger, C.R.; et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008, 4, e1000041. [Google Scholar] [CrossRef]
- Hüffmeier, U.; Uebe, S.; Ekici, A.B.; Bowes, J.; Giardina, E.; Korendowych, E.; Juneblad, K.; Apel, M.; McManus, R.; Ho, P.; et al. Common variants at TRAF3IP2 are asociated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010, 42, 996–999. [Google Scholar] [CrossRef]
- Ellinghaus, E.; Stuart, P.E.; Ellinghaus, D.; Nair, R.P.; Debrus, S.; Raelson, J.V.; Belouchi, M.; Tejasvi, T.; Li, Y.; Tsoi, L.C.; et al. Genome-wide meta-analysis of psoriatic arthritis identifies susceptibility locus at REL. J. Investig. Dermatol. 2012, 132, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yuan, F.; Fan, X.; Wang, Y. Psoriatic arthritis: A systematic review of non-HLA genetic studies and important signaling pathways. Int. J. Rheum. Dis. 2020. online. [Google Scholar] [CrossRef] [PubMed]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, eaay5012. [Google Scholar] [CrossRef] [PubMed]
- Mills, M.C.; Rahal, C. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat. Genet. 2020, 52, 242–243. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Génin, E. Missing heritability of complex diseases: Case solved? Hum. Genet. 2020, 139, 103–113. [Google Scholar] [CrossRef]
- Robinson, P.C.; Leo, P.J.; Pointon, J.J.; Harris, J.; Cremin, K.; Bradbury, L.A.; Stebbings, S.; Harrison, A.A.; Duncan, E.L.; Evans, D.M.; et al. Exome-wide study of ankylosing spondylitis demonstrates additional shared genetic background with inflammatory bowel disease. NPJ Genom. Med. 2016, 1, 16008. [Google Scholar] [CrossRef]
- O’Rielly, D.D.; Uddin, M.; Codner, D.; Hayley, M.; Zhou, J.; Pena-Castillo, L.; Mostafa, A.A.; Hasan, S.M.M.; Liu, W.; Haroon, N.; et al. Private rare deletions in SEC16A and MAMDC4 may represent novel pathogenic variants in familial axial spondyloarthritis. Ann. Rheum. Dis. 2016, 75, 772–779. [Google Scholar] [CrossRef]
- Rong, J.; Li, Q.; Zhang, P.; Wu, X.; Huang, J.; Li, C.; Liao, Z.; Xie, Y.; Lv, Q.; Wei, Q.; et al. A rare co-segregation-mutation in the insulin receptor substrate 1 gene in one Chinese family with ankylosing spondylitis. PLoS ONE 2015, 10, e0126348. [Google Scholar] [CrossRef]
- Tan, Z.; Zeng, H.; Xu, Z.; Tian, Q.; Gao, X.; Zhou, C.; Zheng, Y.; Wang, J.; Ling, G.; Wang, B.; et al. Identification of ANKDD1B variants in an ankylosing spondylitis pedigree and a sporadic patient. BMC Med. Genet. 2018, 19, 111. [Google Scholar] [CrossRef]
- Feng, Y.; Hong, Y.; Zhang, X.; Cao, C.; Yang, X.; Lai, S.; Fan, C.; Cheng, F.; Yan, M.; Li, C.; et al. Genetic variants of TREML2 are associated with HLA-B27-positive ankylosing spondylitis. Gene 2018, 668, 121–128. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef]
- Uddin, M.; Maksymowych, W.P.; Inman, R.; Gladman, D.; Munn, A.; Yazdani, R.; Pellett, F.; Hamilton, S.; O’Rielly, D.D.; Rahman, P. UGT2B17 copy number gain in a large ankylosing spondylitis multiplex family. BMC Genet. 2013, 14, 67. [Google Scholar] [CrossRef]
- Jung, S.H.; Yim, S.H.; Hu, H.J.; Lee, K.H.; Lee, J.H.; Sheen, D.H.; Lim, M.K.; Kim, S.Y.; Park, S.W.; Kim, S.H.; et al. Genome-wide copy number variation analysis identifies deletion variants associated with ankylosing spondylitis. Arthritis Rheumatol. 2014, 66, 2103–2112. [Google Scholar] [CrossRef]
- Wei, W.H.; Hemani, G.; Haley, C.S. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef]
- Kirino, Y.; Bertsias, G.; Ishigatsubo, Y.; Mizuki, N.; Tugal-Tutkun, I.; Seyahi, E.; Ozyazgan, Y.; Sacli, F.S.; Erer, B.; Inoko, H.; et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat. Genet. 2013, 45, 202–207. [Google Scholar] [CrossRef]
- Genetic Analysis of Psoriasis Consortium; The Wellcome Trust Case Control Consortium 2; Strange, A.; Capon, F.; Spencer, C.C.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.; et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [Google Scholar]
- Castro-Santos, P.; Moro-García, M.A.; Marcos-Fernández, R.; Alonso-Arias, R.; Díaz-Peña, R. ERAP1 and HLA-C interaction in inflammatory bowel disease in the Spanish population. Innate Immun. 2017, 23, 476–481. [Google Scholar] [CrossRef]
- Caulfield, T.; Fullerton, S.M.; Ali-Khan, S.E.; Arbour, L.; Burchard, E.G.; Cooper, R.S.; Hardy, B.J.; Harry, S.; Hyde-Lay, R.; Kahn, J.; et al. Race and ancestry in biomedical research: Exploring the challenges. Genome Med. 2009, 1, 1–8. [Google Scholar] [CrossRef]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef]
- Loh, P.R.; Bhatia, G.; Gusev, A.; Finucane, H.K.; Bulik-Sullivan, B.K.; Pollack, S.J.; De Candia, T.R.; Lee, S.H.; Wray, N.R.; Kendler, K.S.; et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015, 47, 1385–1392. [Google Scholar] [CrossRef]
- Jansen, P.R.; Watanabe, K.; Stringer, S.; Skene, N.; Bryois, J.; Hammerschlag, A.R.; de Leeuw, C.A.; Benjamins, J.S.; Muñoz-Manchado, A.B.; Nagel, M.; et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet. 2019, 51, 394–403. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Brown, M.A.; Li, Z.; Cao, K.A.L. Biomarker development for axial spondyloarthritis. Nat. Rev. Rheumatol. 2020, 16, 448–463. [Google Scholar] [CrossRef]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Rostami, S.; Hoff, M.; Brown, M.A.; Hveem, K.; Holmen, O.L.; Fritsche, L.G.; Videm, V. Prediction of ankylosing spondylitis in the HUNT study by a genetic risk score combining 110 single-nucleotide polymorphisms of genome-wide significance. J. Rheumatol. 2020, 47, 204–210. [Google Scholar] [CrossRef]
- Knevel, R.; Cessie, S.L.; Terao, C.C.; Slowikowski, K.; Cui, J.; Huizinga, T.W.J.; Costenbader, K.H.; Liao, K.P.; Karlson, E.W.; Raychaudhuri, S. Using genetics to prioritize diagnoses for rheumatology outpatients with inflammatory arthritis. Sci. Transl. Med. 2020, 12, eaay1548. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Participants, Main Assessments | Conclusions |
|---|---|---|
| [53] | 366 patients with PsA and 299 controls. KIR and HLA genotyping. | The activating KIR2DS1 and/or KIR2DS2 genes were more frequent in patients, showing their association with disease risk, but only when HLA ligands for their homologous inhibitory receptors, KIR2DL1 and KIR2DL2/3, were missing. |
| [54] | 220 patients positive for psoriasis vulgaris, 75 also diagnosed as positive for PsA, and 90 controls. KIR genotyping. | The activating KIR2DS1 gene was more frequent in patients with PsA, compared to those with psoriasis negative for PsA, and to unaffected controls. |
| [55] | 396 patients with psoriasis and 372 controls. Psoriasis vulgaris without joint symptoms was diagnosed in 241 patients, guttate (or ‘eruptive’) psoriasis in 80 patients, and PsA in 75 patients. KIR and HLA-C genotyping. | There was a trend towards a higher KIR2DS1 gene frequency among patients with PsA. |
| [56] | Two HLA-B27–positive Caucasian populations were selected (Spain: 71 patients with AS and 105 controls; and Azores, Portugal: 55 patients with AS and 75 controls). HLA-B and KIR3DS1/3DL1 genotyping | The activating KIR3DS1 gene was associated with AS compared with B27 controls, whereas the inhibitory KIR3DL1 gene was decreased in patients with AS compared with B27 controls. The effect of KIR3DL1 (protection) or KIR3DS1 gene (susceptibility) on AS might stronger when the corresponding ligand Bw4-I80 is present. |
| [57] | Two HLA-B27–positive Asian populations were selected (China: 42 patients with AS and 30 controls; and Thailand: 30 patients with AS and 16 controls) | KIR3DS1, KIR2DS5, and KIR2DL5 genes were more frequent in patients with AS. The frequency of 3DL1/3DL1 and 3DL1/3DS1 genotypes was lower and higher in patients, respectively. |
| [58] | 200 patients with AS and 405 controls. KIR genotyping. | No differences in KIR genotype frequencies between patients and controls. |
| [59] | 83 patients with AS and 107 controls, all HLA-B27–positive. KIR3DL1 and KIR3DS1 subtyping (genes and alleles). | The frequency of the inhibitory KIR3DL1 gene was lower in patients than in B27 controls. The KIR3DL1 gene was negatively associated with AS at the expense of alleles encoding functional receptors (KIR3DL1*F) but not of KIR3DL1*004 (not functional). |
| [60] | 115 patients with AS and 119 controls, all HLA-B27–positive. KIR and HLA-C genotyping. KIR and HLA-C genotyping. | The frequency of the inhibitory KIR2DL1 and KIR2DL5 gene was higher in patients than in controls. |
| [61] | 270 patients with AS and 435 controls, all HLA-B27–positive. KIR3DL1 and KIR3DS1 subtyping (genes and alleles), and HLA-B genotyping. | The activating KIR3DS1*013 allele was more frequent in patients independent of the presence of the HLA-Bw4I80 epitope, whereas the presence of inhibitory allotypes such as KIR3DL1*004 demonstrated a negative association in patients in the presence of HLA-Bw4I80. |
| [62] | 35 patients with AS and 200 controls. KIR and HLA genotyping. | The telomeric KIR2DL5A, KIR2DS1, and KIR3DS1 genes were more frequent in patients compared to controls. KIR3DL1/Bw4I80 and KIR2DS1/C2 compound genotypes showed association with AS. |
| [63] | 60 patients with AS and 60 controls. KIR genotyping. | The activating KIR3DS1 gene was more frequent in patients than in controls. The frequency of KIR3DL1/KIR3DL1 genotype was lower in patients than in controls. |
| [64] | 110 patients with AS, 86 patients with Behçet disease, and 154 controls. KIR genotyping. | Compared with controls, the frequency of the inhibitory gene KIR3DL1 was lower in patients with AS and also in those affected by uveitis. |
| [65] | 678 patients with PsA and 688 controls. KIR and HLA genotyping. | The activating KIR2DS2 gene was more frequent in patients than in controls. |
| [66] | 176 patients with AS and 435 controls. KIR genotyping. | The frequency of the KIR2DS1 and KIR3DS1 genes was higher in patients than in controls. |
| [67] | 200 patients with PsA and 200 controls. KIR and HLA genotyping. | The inhibitory KIR2DL3 gene was more frequent in patients than in controls, whereas the frequency of the inhibitory KIR2DL5 gene was lower. |
| [68] | Successful genotyping in 392 patients with PsA, 260 patients with cutaneous psoriasis and with no arthritis, and 371 controls. KIR3DL1 subtyping (alleles). | The non-functional KIR3DL1 allele was associated with psoriatic diseases (i.e., higher frequency in both types of patients compared to controls). |
| [69] | 653 patients with AS and 952 controls. KIR and HLA-C genotyping. | The frequency of the inhibitory KIR2DL5 gene was lower in patients. In addition, KIR2DL5 combined with the HLA-C1/C2 heterozygous genotype showed a protective effect against AS. |
| [70] | 138 patients with ReA and 151 controls. KIR and HLA-C genotyping. | The inhibitory KIR2DL2 and KIR2DL5 genes were less frequent in patients than in controls. The activating KIR2DS1 alone or in combination with the HLA-C1C1 genotype was associated with susceptibility to ReA, whereas KIR2DL2 in combination with the HLA-C1 ligand was associated with protection against ReA. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Peña, R.; Castro-Santos, P.; Durán, J.; Santiago, C.; Lucia, A. The Genetics of Spondyloarthritis. J. Pers. Med. 2020, 10, 151. https://doi.org/10.3390/jpm10040151
Díaz-Peña R, Castro-Santos P, Durán J, Santiago C, Lucia A. The Genetics of Spondyloarthritis. Journal of Personalized Medicine. 2020; 10(4):151. https://doi.org/10.3390/jpm10040151
Chicago/Turabian StyleDíaz-Peña, Roberto, Patricia Castro-Santos, Josefina Durán, Catalina Santiago, and Alejandro Lucia. 2020. "The Genetics of Spondyloarthritis" Journal of Personalized Medicine 10, no. 4: 151. https://doi.org/10.3390/jpm10040151
APA StyleDíaz-Peña, R., Castro-Santos, P., Durán, J., Santiago, C., & Lucia, A. (2020). The Genetics of Spondyloarthritis. Journal of Personalized Medicine, 10(4), 151. https://doi.org/10.3390/jpm10040151