Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation

{kind=link}
Abstract
1. Introduction
2. Clinical Characteristics
2.1. Ocular Manifestations
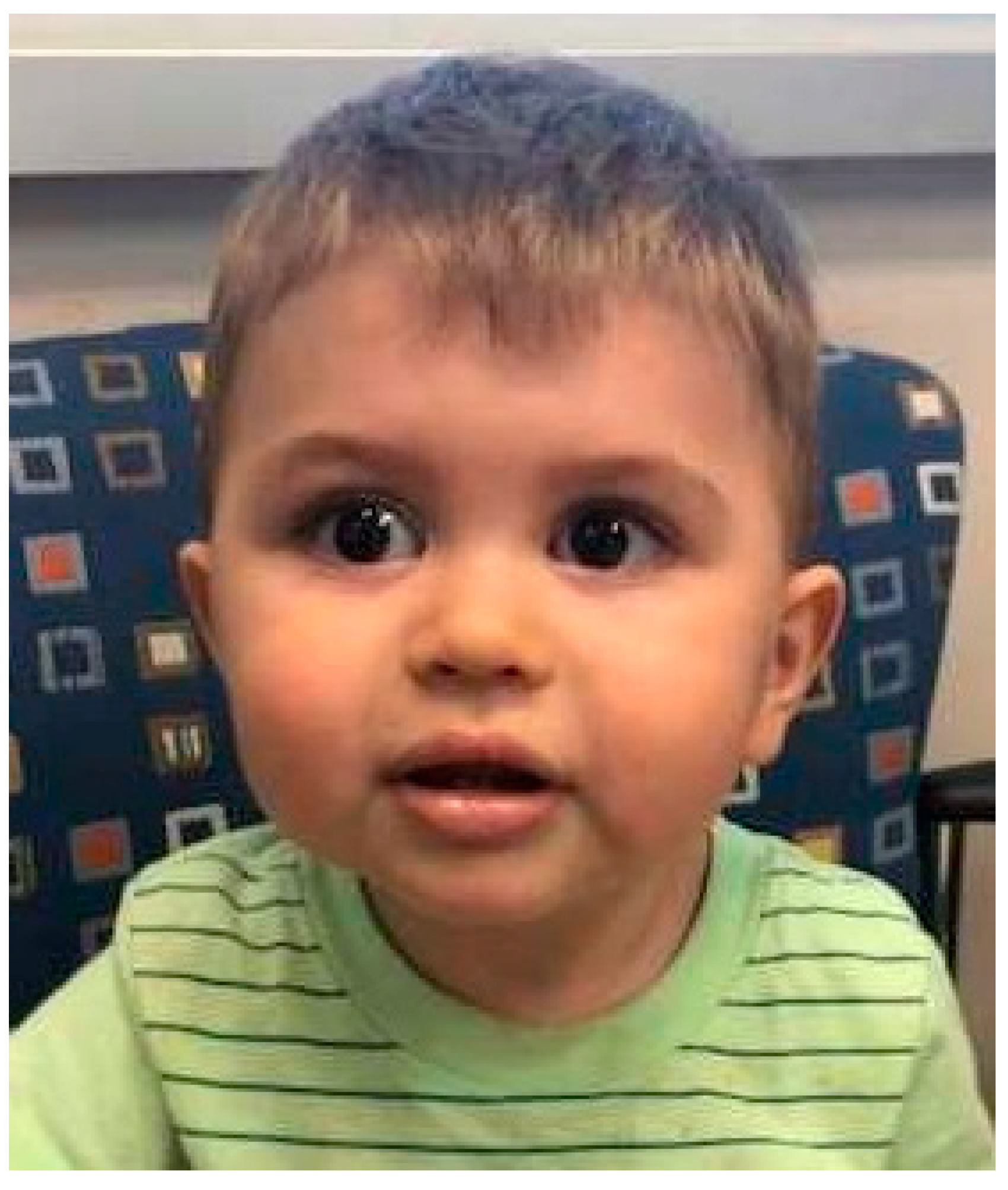
2.2. Craniofacial Manifestations
2.3. Auditory Manifestations
2.4. Skeletal Manifestations
3. Diagnosing Stickler Syndrome
3.1. Chromosomal Microarray
3.2. Gene Testing
3.3. Exome and Genome Sequencing
3.4. Interpreting Genetic Testing Results
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stickler, G.B.; Belau, P.G.; Farrell, F.J.; Jones, J.D.; Pugh, D.G.; Steinberg, A.G.; Ward, L.E. Hereditary Progressive Arthro-Ophthalmopathy. Mayo Clin. Proc. 1965, 40, 433–455. [Google Scholar] [PubMed]
- Snead, M.P.; Yates, J.R. Clinical and Molecular genetics of Stickler syndrome. J. Med. Genet. 1999, 36, 353–359. [Google Scholar] [PubMed]
- Snead, M.P.; McNinch, A.M.; Poulson, A.V.; Bearcroft, P.; Silverman, B.; Gomersall, P.; Parfect, V.; Richards, A.J. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye 2011, 25, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.J.; Blair, M.P.; Solinski, M.A.; Zhang, D.L.; Jabbehdari, S. The importance of early diagnosis of Stickler syndrome: Finding opportunities for preventing blindness. Taiwan J. Ophthalmol. 2018, 8, 189–195. [Google Scholar] [CrossRef]
- Stickler, G.B.; Hughes, W.; Houchin, P. Clinical features of hereditary progressive arthro-ophthalmopathy (Stickler syndrome): A survey. Genet. Med. 2001, 3, 192–196. [Google Scholar] [CrossRef]
- Robin, N.H.; Moran, R.T.; Ala-Kokko, L. Stickler Syndrome. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20301479 (accessed on 20 May 2020).
- Annunen, S.; Korkko, J.; Czarny, M.; Warman, M.L.; Brunner, H.G.; Kaariainen, H.; Mulliken, J.B.; Tranebjaerg, L.; Brooks, D.G.; Cox, G.F.; et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am. J. Hum. Genet. 1999, 65, 974–983. [Google Scholar] [CrossRef]
- Acke, F.R.; Malfait, F.; Vanakker, O.M.; Steyaert, W.; De Leeneer, K.; Mortier, G.; Dhooge, I.; De Paepe, A.; De Leenheer, E.M.; Coucke, P.J. Novel pathogenic COL11A1/COL11A2 variants in Stickler syndrome detected by targeted NGS and exome sequencing. Mol. Genet. Metab. 2014, 113, 230–235. [Google Scholar] [CrossRef]
- Van Camp, G.; Snoeckx, R.L.; Hilgert, N.; van den Ende, J.; Fukuoka, H.; Wagatsuma, M.; Suzuki, H.; Smets, R.M.; Vanhoenacker, F.; Declau, F.; et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am. J. Hum. Genet. 2006, 79, 449–457. [Google Scholar] [CrossRef]
- Baker, S.; Booth, C.; Fillman, C.; Shapiro, M.; Blair, M.P.; Hyland, J.C.; Ala-Kokko, L. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am. J. Med. Genet. A 2011, 155A, 1668–1672. [Google Scholar] [CrossRef]
- Faletra, F.; D’Adamo, A.P.; Bruno, I.; Athanasakis, E.; Biskup, S.; Esposito, L.; Gasparini, P. Autosomal recessive Stickler syndrome due to a loss of function mutation in the COL9A3 gene. Am. J. Med. Genet. A 2014, 164, 42–47. [Google Scholar] [CrossRef]
- Ahmad, N.N.; Ala-Kokko, L.; Knowlton, R.G.; Jimenez, S.A.; Weaver, E.J.; Maguire, J.I.; Tasman, W.; Prockop, D.J. Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc. Natl. Acad. Sci. USA 1991, 88, 6624–6627. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.J.; Yates, J.R.; Williams, R.; Payne, S.J.; Pope, F.M.; Scott, J.D.; Snead, M.P. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum. Mol. Genet. 1996, 5, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Sirko-Osadsa, D.A.; Murray, M.A.; Scott, J.A.; Lavery, M.A.; Warman, M.L.; Robin, N.H. Stickler syndrome without eye involvement is caused by mutations in COL11A2, the gene encoding the alpha2(XI) chain of type XI collagen. J. Pediatr. 1998, 132, 368–371. [Google Scholar] [CrossRef]
- Hoornaert, K.P.; Vereecke, I.; Dewinter, C.; Rosenberg, T.; Beemer, F.A.; Leroy, J.G.; Bendix, L.; Bjorck, E.; Bonduelle, M.; Boute, O.; et al. Stickler syndrome caused by COL2A1 mutations: Genotype-phenotype correlation in a series of 100 patients. Eur. J. Hum. Genet. 2010, 18, 872–880. [Google Scholar] [CrossRef]
- Coussa, R.G.; Sears, J.; Traboulsi, E.I. Stickler syndrome: Exploring prophylaxis for retinal detachment. Curr. Opin. Ophthalmol. 2019, 30, 306–313. [Google Scholar] [CrossRef]
- Ang, A.; Poulson, A.V.; Goodburn, S.F.; Richards, A.J.; Scott, J.D.; Snead, M.P. Retinal detachment and prophylaxis in type 1 Stickler syndrome. Ophthalmology 2008, 115, 164–168. [Google Scholar] [CrossRef]
- Donoso, L.A.; Edwards, A.O.; Frost, A.T.; Ritter, R., 3rd; Ahmad, N.; Vrabec, T.; Rogers, J.; Meyer, D.; Parma, S. Clinical variability of Stickler syndrome: Role of exon 2 of the collagen COL2A1 gene. Surv. Ophthalmol. 2003, 48, 191–203. [Google Scholar] [CrossRef]
- McAlinden, A.; Majava, M.; Bishop, P.N.; Perveen, R.; Black, G.C.; Pierpont, M.E.; Ala-Kokko, L.; Mannikko, M. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of Stickler syndrome. Hum. Mutat. 2008, 29, 83–90. [Google Scholar] [CrossRef]
- Richards, A.J.; Meredith, S.; Poulson, A.; Bearcroft, P.; Crossland, G.; Baguley, D.M.; Scott, J.D.; Snead, M.P. A novel mutation of COL2A1 resulting in dominantly inherited rhegmatogenous retinal detachment. Investig. Ophthalmol. Vis. Sci. 2005, 46, 663–668. [Google Scholar] [CrossRef]
- Go, S.L.; Maugeri, A.; Mulder, J.J.; van Driel, M.A.; Cremers, F.P.; Hoyng, C.B. Autosomal dominant rhegmatogenous retinal detachment associated with an Arg453Ter mutation in the COL2A1 gene. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4035–4043. [Google Scholar] [CrossRef]
- Richards, A.J.; McNinch, A.; Martin, H.; Oakhill, K.; Rai, H.; Waller, S.; Treacy, B.; Whittaker, J.; Meredith, S.; Poulson, A.; et al. Stickler syndrome and the vitreous phenotype: Mutations in COL2A1 and COL11A1. Hum. Mutat. 2010, 31, E1461–E1471. [Google Scholar] [CrossRef] [PubMed]
- Hanson-Kahn, A.; Li, B.; Cohn, D.H.; Nickerson, D.A.; Bamshad, M.J.; University of Washington Center for Mendelian, G.; Hudgins, L. Autosomal recessive Stickler syndrome resulting from a COL9A3 mutation. Am. J. Med. Genet. A 2018, 176, 2887–2891. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.J.; Fincham, G.S.; McNinch, A.; Hill, D.; Poulson, A.V.; Castle, B.; Lees, M.M.; Moore, A.T.; Scott, J.D.; Snead, M.P. Alternative splicing modifies the effect of mutations in COL11A1 and results in recessive type 2 Stickler syndrome with profound hearing loss. J. Med. Genet. 2013, 50, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, F.; Al Hazzaa, S.A.; Tayeb, H.; Alkuraya, F.S. LOXL3, encoding lysyl oxidase-like 3, is mutated in a family with autosomal recessive Stickler syndrome. Hum. Genet. 2015, 134, 451–453. [Google Scholar] [CrossRef]
- Chan, T.K.; Alkaabi, M.K.; ElBarky, A.M.; El-Hattab, A.W. LOXL3 novel mutation causing a rare form of autosomal recessive Stickler syndrome. Clin. Genet. 2019, 95, 325–328. [Google Scholar] [CrossRef]
- Schrauwen, I.; Sommen, M.; Claes, C.; Pinner, J.; Flaherty, M.; Collins, F.; Van Camp, G. Broadening the phenotype of LRP2 mutations: A new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clin. Genet. 2014, 86, 282–286. [Google Scholar] [CrossRef]
- Poulson, A.V.; Hooymans, J.M.; Richards, A.J.; Bearcroft, P.; Murthy, R.; Baguley, D.M.; Scott, J.D.; Snead, M.P. Clinical features of type 2 Stickler syndrome. J. Med. Genet. 2004, 41, e107. [Google Scholar] [CrossRef]
- Snead, M.P.; Payne, S.J.; Barton, D.E.; Yates, J.R.; Al-Imara, L.; Pope, F.M.; Scott, J.D. Stickler syndrome: Correlation between vitreoretinal phenotypes and linkage to COL 2A1. Eye 1994, 8 Pt 6, 609–614. [Google Scholar] [CrossRef]
- Scott, J.D. Congenital myopia and retinal detachment. Trans. Ophthalmol. Soc. U. K. 1980, 100, 69–71. [Google Scholar]
- Copikova, J.; Paderova, J.; Romankova, V.; Havlovicova, M.; Balascakova, M.; Zelinova, M.; Vejvalkova, S.; Simandlova, M.; Stepankova, J.; Horinova, V.; et al. Expanding the phenotype spectrum associated with pathogenic variants in the COL2A1 and COL11A1 genes. Ann. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Seery, C.M.; Pruett, R.C.; Liberfarb, R.M.; Cohen, B.Z. Distinctive cataract in the Stickler syndrome. Am. J. Ophthalmol. 1990, 110, 143–148. [Google Scholar] [CrossRef]
- Nielsen, C.E. Stickler’s syndrome. Acta Ophthalmol. (Copenh) 1981, 59, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Nixon, T.R.W.; Alexander, P.; Richards, A.; McNinch, A.; Bearcroft, P.W.P.; Cobben, J.; Snead, M.P. Homozygous Type IX collagen variants (COL9A1, COL9A2, and COL9A3) causing recessive Stickler syndrome-Expanding the phenotype. Am. J. Med. Genet. A 2019, 179, 1498–1506. [Google Scholar] [CrossRef]
- Mossey, P.A.; Little, J.; Munger, R.G.; Dixon, M.J.; Shaw, W.C. Cleft lip and palate. Lancet 2009, 374, 1773–1785. [Google Scholar] [CrossRef]
- Acke, F.R.; Dhooge, I.J.; Malfait, F.; De Leenheer, E.M. Hearing impairment in Stickler syndrome: A systematic review. Orphanet J. Rare Dis. 2012, 7, 84. [Google Scholar] [CrossRef]
- Acke, F.R.; Swinnen, F.K.; Malfait, F.; Dhooge, I.J.; De Leenheer, E.M. Auditory phenotype in Stickler syndrome: Results of audiometric analysis in 20 patients. Eur. Arch. Otorhinolaryngol. 2016, 273, 3025–3034. [Google Scholar] [CrossRef]
- Szymko-Bennett, Y.M.; Mastroianni, M.A.; Shotland, L.I.; Davis, J.; Ondrey, F.G.; Balog, J.Z.; Rudy, S.F.; McCullagh, L.; Levy, H.P.; Liberfarb, R.M.; et al. Auditory dysfunction in Stickler syndrome. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1061–1068. [Google Scholar] [CrossRef][Green Version]
- Rose, P.S.; Levy, H.P.; Liberfarb, R.M.; Davis, J.; Szymko-Bennett, Y.; Rubin, B.I.; Tsilou, E.; Griffith, A.J.; Francomano, C.A. Stickler syndrome: Clinical characteristics and diagnostic criteria. Am. J. Med. Genet. A 2005, 138A, 199–207. [Google Scholar] [CrossRef]
- Rose, P.S.; Ahn, N.U.; Levy, H.P.; Ahn, U.M.; Davis, J.; Liberfarb, R.M.; Nallamshetty, L.; Sponseller, P.D.; Francomano, C.A. Thoracolumbar spinal abnormalities in Stickler syndrome. Spine 2001, 26, 403–409. [Google Scholar] [CrossRef]
- ACMG Board of Directors. Clinical utility of genetic and genomic services: A position statement of the American College of Medical Genetics and Genomics. Genet. Med. 2015, 17, 505–507. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- South, S.T.; Lee, C.; Lamb, A.N.; Higgins, A.W.; Kearney, H.M.; Working Group for the American College of Medical, G.; Genomics Laboratory Quality Assurance, C. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet. Med. 2013, 15, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Richards, A.J.; McNinch, A.; Whittaker, J.; Treacy, B.; Oakhill, K.; Poulson, A.; Snead, M.P. Splicing analysis of unclassified variants in COL2A1 and COL11A1 identifies deep intronic pathogenic mutations. Eur. J. Hum. Genet. 2012, 20, 552–558. [Google Scholar] [CrossRef][Green Version]
- Sun, W.; Xiao, X.; Li, S.; Jia, X.; Zhang, Q. A novel deep intronic COL2A1 mutation in a family with early-onset high myopia/ocular-only Stickler syndrome. Ophthalmic Physiol. Opt. 2020, 40, 281–288. [Google Scholar] [CrossRef]
- Shashi, V.; McConkie-Rosell, A.; Rosell, B.; Schoch, K.; Vellore, K.; McDonald, M.; Jiang, Y.H.; Xie, P.; Need, A.; Goldstein, D.B. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2014, 16, 176–182. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Biesecker, B.B. An approach to pediatric exome and genome sequencing. Curr. Opin. Pediatr. 2014, 26, 639–645. [Google Scholar] [CrossRef]
- ACMG Board of Directors. Points to consider for informed consent for genome/exome sequencing. Genet. Med. 2013, 15, 748–749. [Google Scholar] [CrossRef] [PubMed]
- ACMG Board of Directors. Points to consider in the clinical application of genomic sequencing. Genet. Med. 2012, 14, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boothe, M.; Morris, R.; Robin, N. Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation. J. Pers. Med. 2020, 10, 105. https://doi.org/10.3390/jpm10030105
Boothe M, Morris R, Robin N. Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation. Journal of Personalized Medicine. 2020; 10(3):105. https://doi.org/10.3390/jpm10030105
Chicago/Turabian StyleBoothe, Megan, Robert Morris, and Nathaniel Robin. 2020. "Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation" Journal of Personalized Medicine 10, no. 3: 105. https://doi.org/10.3390/jpm10030105
APA StyleBoothe, M., Morris, R., & Robin, N. (2020). Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation. Journal of Personalized Medicine, 10(3), 105. https://doi.org/10.3390/jpm10030105