Inhaled Xenon Washout as a Biomarker of Alzheimer’s Disease


Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval and Consent to Participate
2.2. Participant Recruitment
2.3. 1H Magnetic Resonance Imaging
2.4. 129Xe Magnetic Resonance Spectroscopy
2.5. 129Xe Magnetic Resonance Imaging
2.6. 129Xe Image Processing
2.7. Xenon Washout Parameter Maps Image Processing
2.8. Statistical Analysis
3. Results
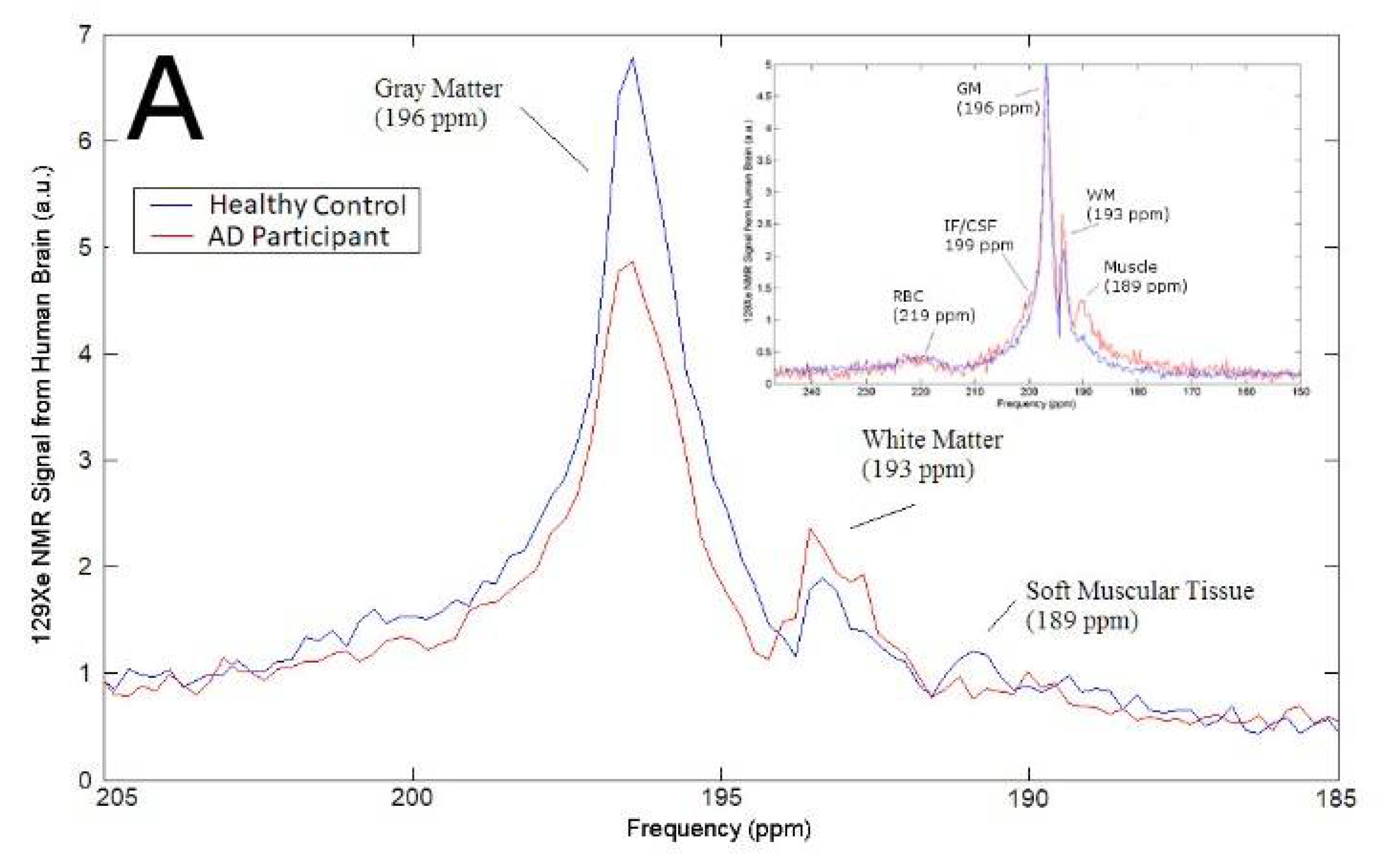
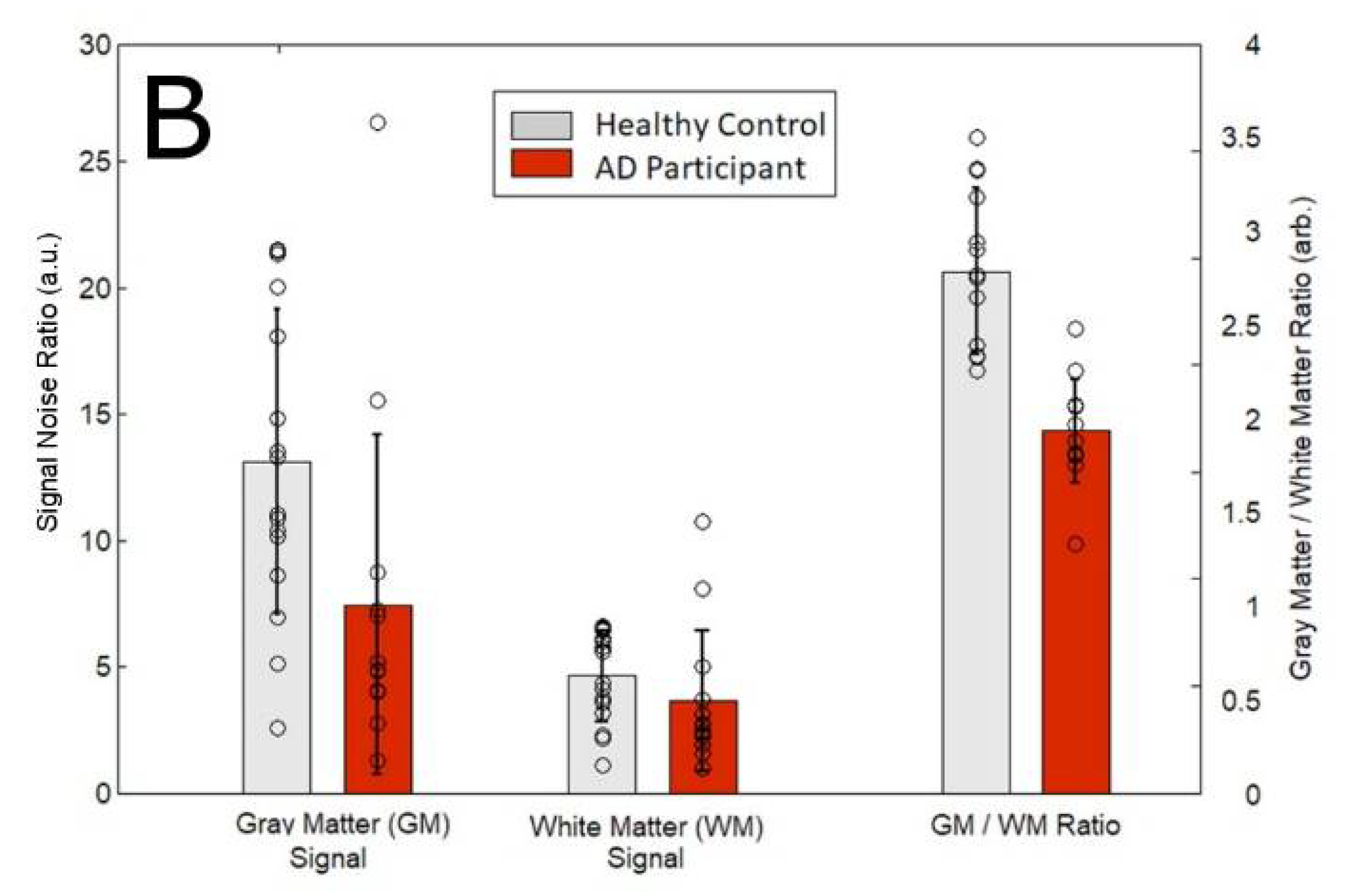
3.1. Magnetic Resonance Spectroscopy
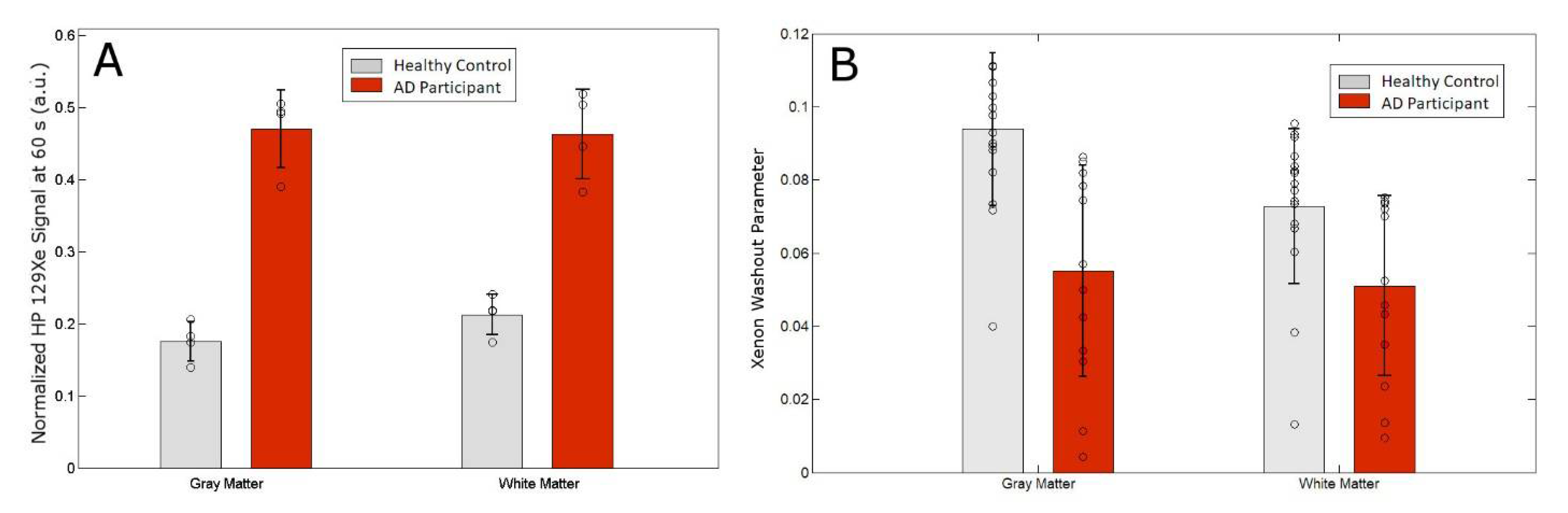
3.2. MRS as a Function of Time
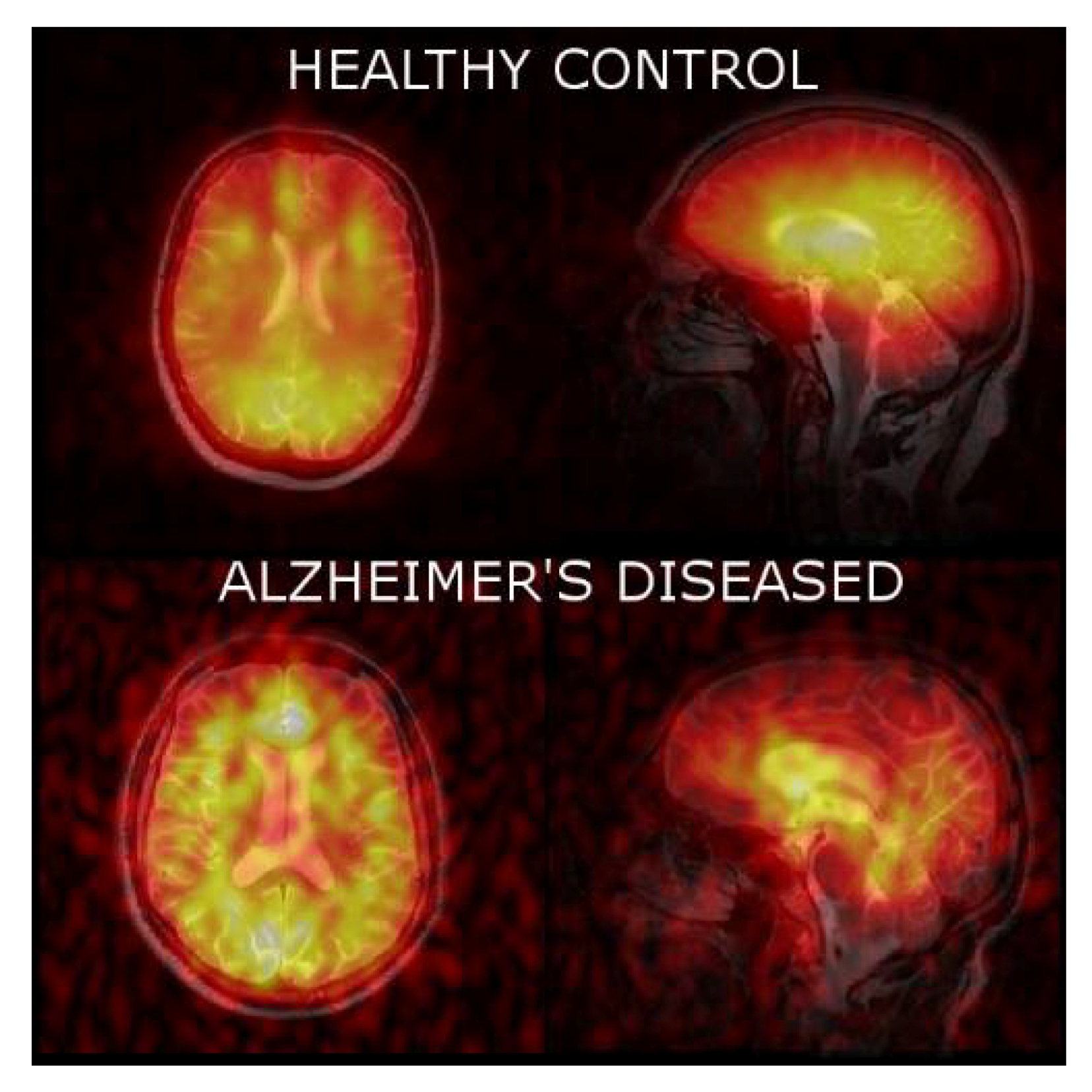
3.3. 129Xe MRI
4. Discussion
Supplementary Materials
Author Contribution
Funding
Acknowledgments
Conflict of Interest
Trial Registration
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Robinson, M.; Lee, B.Y.; Hane, F.T. Recent Progess in Alzheimer’s disease Research, Part 2: Genetics and Epidemiology. J. Alzheimers Dis. 2017, 57, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Medway, C.; Morgan, K. Review: The genetics of Alzheimer’s disease; putting flesh on the bones. Neuropathol. Appl. Neurobiol. 2014, 40, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, F.K.; Al-janabi, T.; Hardy, J.; Karmiloff-smith, A.; Nizetic, D.; Tybulewicz, V.L.J.; Fisher, E.M.C.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling Amyloid-β, Tau, and Metals in Alzheimer’s Disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.; Leonenko, Z. Effect of metals on kinetic pathways of amyloid-β aggregation. Biomolecules 2014, 4, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Lee, B.Y.; Leonenko, Z. Recent Progress in Alzheimer’s Disease Research, Part 1: Pathology. J. Alzheimers Dis. 2017, 57, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F.X. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.R.; Leuzy, A.; Benedet, A.L.; Breitner, J.; Gauthier, S.; Rosa-Neto, P. Tracking neuroinflammation in Alzheimer’s disease: The role of positron emission tomography imaging. J. Neuroinflamm. 2014, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid-β protein. Ann. N. Y. Acad. Sci. 2006, 924, 17–25. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Selkoe, D. Aβ oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; George, D.R.; D’Alton, S. Describing the dying days of “Alzheimer’s disease”. J. Alzheimers Dis. 2011, 24, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Is the Alzheimer’s Amyloid Hypothesis Wrong? Atlantic 2017. Available online: https://www.theatlantic.com/health/archive/2017/02/alzheimers-amyloid-hypothesis/517185/ (accessed on 5 June 2018).
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Rauk, A. Alzheimer’s disease and the “ABSENT” hypothesis: Mechanism for amyloid beta endothelial and neuronal toxicity. Med. Hypotheses 2005, 65, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Are Major Dementias Triggered by Poor Blood Flow to the Brain? Theoretical Considerations. J. Alzheimers Dis. 2017, 57, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Chi, N.-F.; Chien, L.-N.; Ku, H.-L.; Hu, C.-J.; Chiou, H.-Y. Alzheimer disease and risk of stroke: A population-based cohort study. Neurology 2013, 80, 705–711. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging Brain Amyloid in Alzheimer’s Disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Jack, C.R.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Walhovd, K.B.; Fjell, A.M.; Brewer, J.; McEvoy, L.K.; Fennema-Notestine, C.; Hagler, D.J.; Jennings, R.G.; Karow, D.; Dale, A.M. Alzheimer’s Disease Neuroimaging Initiative Combining MR imaging, positron-emission tomography, and CSF biomarkers in the diagnosis and prognosis of Alzheimer disease. Am. J. Neuroradiol. 2010, 31, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Brys, M.; Pirraglia, E.; Rich, K.; Rolstad, S.; Mosconi, L.; Switalski, R.; Glodzik-Sobanska, L.; De Santi, S.; Zinkowski, R.; Mehta, P.; et al. Prediction and longitudinal study of CSF biomarkers in mild cognitive impairment. Neurobiol. Aging 2009, 30, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Zetterberg, H.; Hansson, O.; Andreasen, N.; Parnetti, L.; Jonsson, M.; Herukka, S.-K.; Flier, W.M.; van der Blankenstein, M.A.; Ewers, M.; et al. CSF Biomarkers and Incipient Alzheimer Disease in Patients With Mild Cognitive Impairment. JAMA 2009, 302, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Barthel, H.; Gertz, H.-J.; Dresel, S.; Peters, O.; Bartenstein, P.; Buerger, K.; Hiemeyer, F.; Wittemer-Rump, S.M.; Seibyl, J.; Reininger, C.; et al. Cerebral amyloid-β PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: A multicentre phase 2 diagnostic study. Lancet Neurol. 2011, 10, 424–435. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Masters, C.L.; Rowe, C.C. Tau imaging: Early progress and future directions. Lancet Neurol. 2015, 14, 114–124. [Google Scholar] [CrossRef]
- Wang, L.; Benzinger, T.L.; Su, Y.; Christensen, J.; Friedrichsen, K.; Aldea, P.; McConathy, J.; Cairns, N.J.; Fagan, A.M.; Morris, J.C.; et al. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions Between β-Amyloid and Tauopathy. JAMA Neurol. 2016, 63110, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J.; et al. Tau and A-beta imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 338ra66. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Wolk, D.A. MRI cortical thickness biomarker predicts AD-like CSF and cognitive decline in normal adults. Neurology 2012, 78, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Basser, P.; Pierpaoli, C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J. Magn. Reson. 1996, 111, 209–219. [Google Scholar] [CrossRef]
- Pierpaoli, C.; Basser, P. Toward a quantitative assessment of diffusion anisotropy. Magn. Reson. Med. 1996, 36, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Groves, A.R.; Tamnes, C.K.; Tjelta, L.; Duff, E.P.; Engvig, A.; Walhovd, K.B.; James, A.; Gass, A.; Monsch, A.U.; et al. A common brain network links development, aging, and vulnerability to disease. Proc. Natl. Acad. Sci. USA 2014, 111, 17648–17653. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Jbabdi, S.; Behrens, T.E.J.; Menke, R.A.; Gass, A.; Monsch, A.U.; Rao, A.; Whitcher, B.; Kindlmann, G.; Matthews, P.M.; et al. DTI measures in crossing-fibre areas: Increased diffusion anisotropy reveals early white matter alteration in MCI and mild Alzheimer’s disease. Neuroimage 2011, 55, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Robinson, M.; Lee, B.Y.; Bai, O.; Leonenko, Z.; Albert, M.S. Recent Progress in Alzheimer’s Disease Research, Part 3: Diagnosis and Treatment. J. Alzheimers Dis. 2017, 57, 645–665. [Google Scholar] [CrossRef] [PubMed]
- Herholz, K.; Ebmeier, K. Clinical amyloid imaging in Alzheimer’s disease. Lancet Neurol. 2011, 10, 667–670. [Google Scholar] [CrossRef]
- Teipel, S.; Drzezga, A.; Grothe, M.J.; Barthel, H.; Chételat, G.; Schuff, N.; Skudlarski, P.; Cavedo, E.; Frisoni, G.B.; Hoffmann, W.; et al. Multimodal imaging in Alzheimer’s disease: Validity and usefulness for early detection. Lancet Neurol. 2015, 14, 1037–1053. [Google Scholar] [CrossRef]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Prohovnik, I.; Mayeux, R.; Sackeim, H.A.; Smith, G.; Stern, Y.; Alderson, P.O. Cerebral perfusion as a diagnostic marker of early Alzheimer’s disease. Neurology 1988, 38, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Alsop, D.C.; Dai, W.; Grossman, M.; Detre, J.A. Arterial Spin Labeling Blood Flow MRI: Its Role in the Early Characterization of Alzheimer’s Disease. J. Alzheimers Dis. 2010, 20, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Alsop, D.C.; Detre, J.A.; Grossman, M. Assessment of cerebral blood flow in Alzheimer’s disease by spin-labeled magnetic resonance imaging. Ann. Neurol. 2000, 47, 93–100. [Google Scholar] [CrossRef]
- Musiek, E.S.; Chen, Y.; Korczykowski, M.; Saboury, B.; Martinez, P.M.; Reddin, J.S.; Alavi, A.; Kimberg, D.Y.; Wolk, D.A.; Juline, P.; et al. Direct comparison of fluorodeoxyglucose positron emission tomography and arterial spin labeling magnetic resonance imaging in Alzheimer’s disease. Alzheimers Dement. 2012, 8, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Perani, D.; Di Piero, V.; Vallar, G.; Cappa, S.; Messa, C.; Bottini, G.; Berti, A.; Passafiume, D.; Scarlato, G.; Gerundini, P. Technetium-99m HM-PAO-SPECT study of regional cerebral perfusion in early Alzheimer’s disease. J. Nucl. Med. 1988, 29, 1507–1514. [Google Scholar] [PubMed]
- Parkes, L.M.; Rashid, W.; Chard, D.T.; Tofts, P.S. Normal cerebral perfusion measurements using arterial spin labeling: Reproducibility, stability, and age and gender effects. Magn. Reson. Med. 2004, 51, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Du, A.T.; Jahng, G.H.; Hayasaka, S.; Kramer, J.H.; Rosen, H.J.; Gorno-Tempini, M.L.; Rankin, K.P.; Miller, B.L.; Weiner, M.W.; Schuff, N. Hypoperfusion in frontotemporal dementia and Alzheimer disease by arterial spin labeling MRI. Neurology 2006, 67, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.A.; Jahng, G.-H.; Weiner, M.W.; Miller, B.L.; Chui, H.C.; Jagust, W.J.; Gorno-Tempini, M.L.; Schuff, N. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: Initial experience. Radiology 2005, 234, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Binnewijzend, M.A.A.; Kuijer, J.P.A.; Benedictus, M.R.; van der Flier, W.M.; Wink, A.M.; Wattjes, M.P.; van Berckel, B.N.M.; Scheltens, P.; Barkhof, F. Cerebral Blood Flow Measured with 3D Pseudocontinuous Arterial Spin-labeling MR Imaging in Alzheimer Disease and Mild Cognitive Impairment: A Marker for Disease Severity. Radiology 2013, 267, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Leijenaar, J.F.; van Maurik, I.S.; Kuijer, J.; van der Flier, W.; Scheltens, P.; Barkhof, F.; Prins, N. Lower cerebral blood flow in subjects with Alzheimer’s dementia, mild cognitive impairment, and subjective cognitive decline using 2D phase-contrast MRI. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2017. [Google Scholar] [CrossRef]
- Black, R.D.; Middleton, H.L.; Cates, G.D.; Cofer, G.P.; Driehuys, B.; Happer, W.; Hedlund, L.W.; Johnson, G.A.; Shattuck, M.D.; Swartz, J.C. In vivo He-3 MR images of guinea pig lungs. Radiology 1996, 199, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; Cates, G.D.; Driehuys, B.; Happer, W.; Saam, B.; Springer, C.S.; Wishnia, A. Biological magnetic resonance imaging using laser-polarized 129Xe. Nature 1994, 370, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; Happer, W. Spin-exchange optical pumping of noble-gas nuclei. Rev. Mod. Phys. 1997, 69, 629–642. [Google Scholar] [CrossRef]
- Happer, W. Optical Pumping. Rev. Mod. Phys. 1972, 44, 169–240. [Google Scholar] [CrossRef]
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; Hane, F.T. Hyperpolarized and Inert Gas MRI: From Technology to Applications in Research and Medicine; Elsevier: New York, NY, USA, 2017; ISBN 9780128036754. [Google Scholar]
- Day, S.E.; Kettunen, M.I.; Gallagher, F.A.; Hu, D.-E.; Lerche, M.; Wolber, J.; Golman, K.; Ardenkjaer-Larsen, J.H.; Brindle, K.M. Detecting tumor response to treatment using hyperpolarized 13C magnetic resonance imaging and spectroscopy. Nat. Med. 2007, 13, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Golman, K.; Ardenkjaer-Larsen, J.H.; Petersson, J.S.; Mansson, S.; Leunbach, I. Molecular imaging with endogenous substances. Proc. Natl. Acad. Sci. USA 2003, 100, 10435–10439. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.; Pike, D.; Coxson, H.; McCormack, D.; Parraga, G. Hyperpolarized (3)He ventilation defects used to predict pulmonary exacerbations in mild to moderate chronic obstructive pulmonary disease. Radiology 2014, 273, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Imai, H.; Kimura, A.; Fujiwara, H.; Rao, M.; Wild, J.M.; Albert, M.S. Chapter 16—Brain Imaging Using Hyperpolarized Xenon MRI, 1st ed.; Elsevier: New York, NY, USA, 2017; ISBN 978-0-12-803675-4. [Google Scholar]
- Rao, M.R.; Stewart, N.J.; Griffiths, P.D.; Norquay, G.; Wild, J.M. Imaging Human Brain Perfusion with Inhaled Hyperpolarized 129Xe MR Imaging. Radiology 2017, 31, 162881. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Stewart, N.J.; Norquay, G.; Griffiths, P.D.; Wild, J.M. High resolution spectroscopy and chemical shift imaging of hyperpolarized 129Xe dissolved in the human brain in vivo at 1.5 tesla. Magn. Reson. Med. 2016, 75, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; Aisen, P.S.; Trojanowski, J.Q.; Shaw, L.M.; Bernstein, M.A.; Petersen, R.C.; Weiner, M.W.; et al. Evidence for ordering of Alzheimer disease biomarkers. Arch. Neurol. 2011, 68, 1526–1235. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C. Articles CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Uttner, I.; Schurig, N.; von Arnim, C.A.F.; Lange-asschenfeldt, C.; Brettschneider, J.; Riepe, M.W.; Tumani, H. Amyloid beta 1–42 in cerebrospinal fluid is associated with cognitive plasticity. Psychiatry Res. 2011, 190, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Kilian, W.; Seifert, F.; Rinneber, G. Dynamic NMR spectroscopy of hyperpolarized 129Xe in human brain analyzed by an uptake model. Magn. Reson. Med. 2004, 51, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Williams, R.; Gao, J.-H.; Nickerson, L.; Xiong, J.; Fox, P. The pharmokinetics of hyperpolarized xenon: Implications for cerebral MRI. J. Magn. Reson. Imag. 1997, 7, 848–854. [Google Scholar] [CrossRef]
- Sachdev, P.S.; Zhuang, L.; Braidy, N.; Wen, W. Is Alzheimer’s a disease of the white matter? Curr. Opin. Psychiatry 2013, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s Disease Amyloid-β Binds Copper and Zinc to Generate an Allosterically Ordered Membrane-penetrating Structure Containing Superoxide Dismutase-like Subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.-Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.; Pettingell, W.; Multhaup, G.; Paradis, M.; Vonsattel, J.; Gusella, J.; Beyreuther, K.; Masters, C.; Tanzi, R. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Drolle, E.; Hane, F.; Lee, B.; Leonenko, Z. Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease. Drug Metab. Rev. 2014, 46, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Yates, E.A.; Legleiter, J.; Montie, H.L.; Jefferson, T. Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front. Neurol. 2013, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.; Yates, E.; Legleiter, J. Amyloid-forming proteins alter the local mechanical properties of lipid membranes. Biochemistry 2013, 52, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.; Drolle, E.; Gaikwad, R.; Faught, E.; Leonenko, Z. Amyloid-beta aggregation on model lipid membranes: An atomic force microscopy study. J. Alzheimers Dis. 2011, 26, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Dante, S.; Hauss, T.; Steitz, R.; Canale, C.; Dencher, N.A. Nanoscale structural and mechanical effects of beta-amyloid (1–42) on polymer cushioned membranes: A combined study by neutron reflectometry and AFM Force Spectroscopy. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in lipid membranes may trigger amyloid toxicity in Alzheimer’s disease. PLoS ONE 2017, 12, e0182194. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Controls (n = 4) | Alzheimer’s Participants (n = 4) | |
|---|---|---|
| Mean Age (years) | 70.0 ± 4.5 | 71.3 ± 6.2 |
| Age of AD Diagnosis | N/A | 64.8 ± 4.3 |
| Sex | 2 males/2 females | 3 males/1 female |
| MoCA Score | 28 ± 1 | 21 ± 3 |
| Norm. Xe-WM signal @ 60 s | 0.200 ± 0.0163 | 0.458 ± 0.0531 |
| Norm. Xe-GM signal @ 60 s | 0.174 ± 0.0252 | 0.465 ± 0.569 |
| Xe Washout Parameter–WM | 0.073 ± 0.021 | 0.051 ± 0.025 |
| Xe Washout Parameter−GM | 0.094 ± 0.021 | 0.055 ± 0.029 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hane, F.T.; Li, T.; Plata, J.-A.; Hassan, A.; Granberg, K.; Albert, M.S. Inhaled Xenon Washout as a Biomarker of Alzheimer’s Disease. Diagnostics 2018, 8, 41. https://doi.org/10.3390/diagnostics8020041
Hane FT, Li T, Plata J-A, Hassan A, Granberg K, Albert MS. Inhaled Xenon Washout as a Biomarker of Alzheimer’s Disease. Diagnostics. 2018; 8(2):41. https://doi.org/10.3390/diagnostics8020041
Chicago/Turabian StyleHane, Francis T., Tao Li, Jennifer-Anne Plata, Ayman Hassan, Karl Granberg, and Mitchell S. Albert. 2018. "Inhaled Xenon Washout as a Biomarker of Alzheimer’s Disease" Diagnostics 8, no. 2: 41. https://doi.org/10.3390/diagnostics8020041
APA StyleHane, F. T., Li, T., Plata, J.-A., Hassan, A., Granberg, K., & Albert, M. S. (2018). Inhaled Xenon Washout as a Biomarker of Alzheimer’s Disease. Diagnostics, 8(2), 41. https://doi.org/10.3390/diagnostics8020041