
Liposclerosing Myxofibrous Tumor: A Separated Clinical Entity?

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
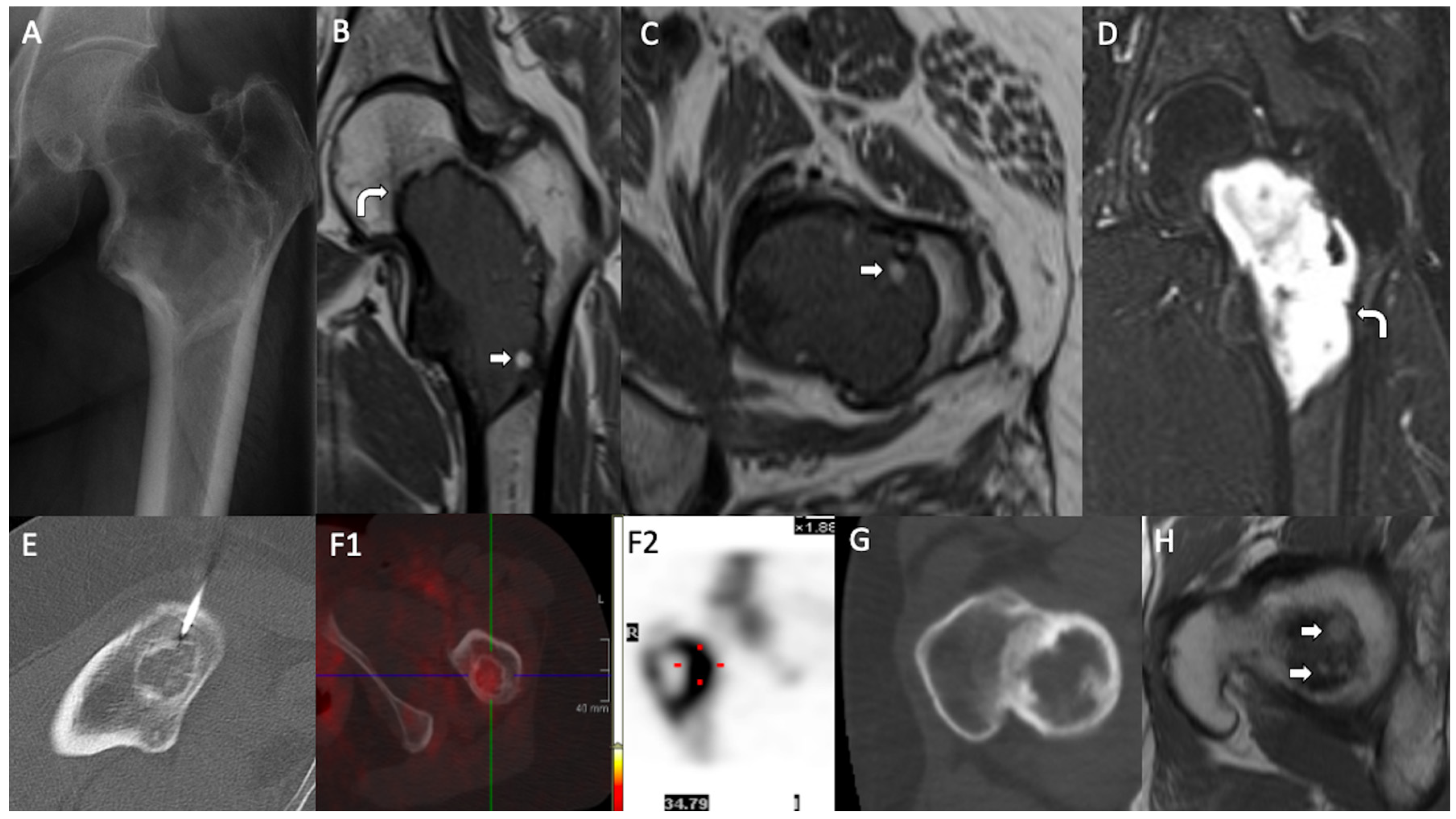
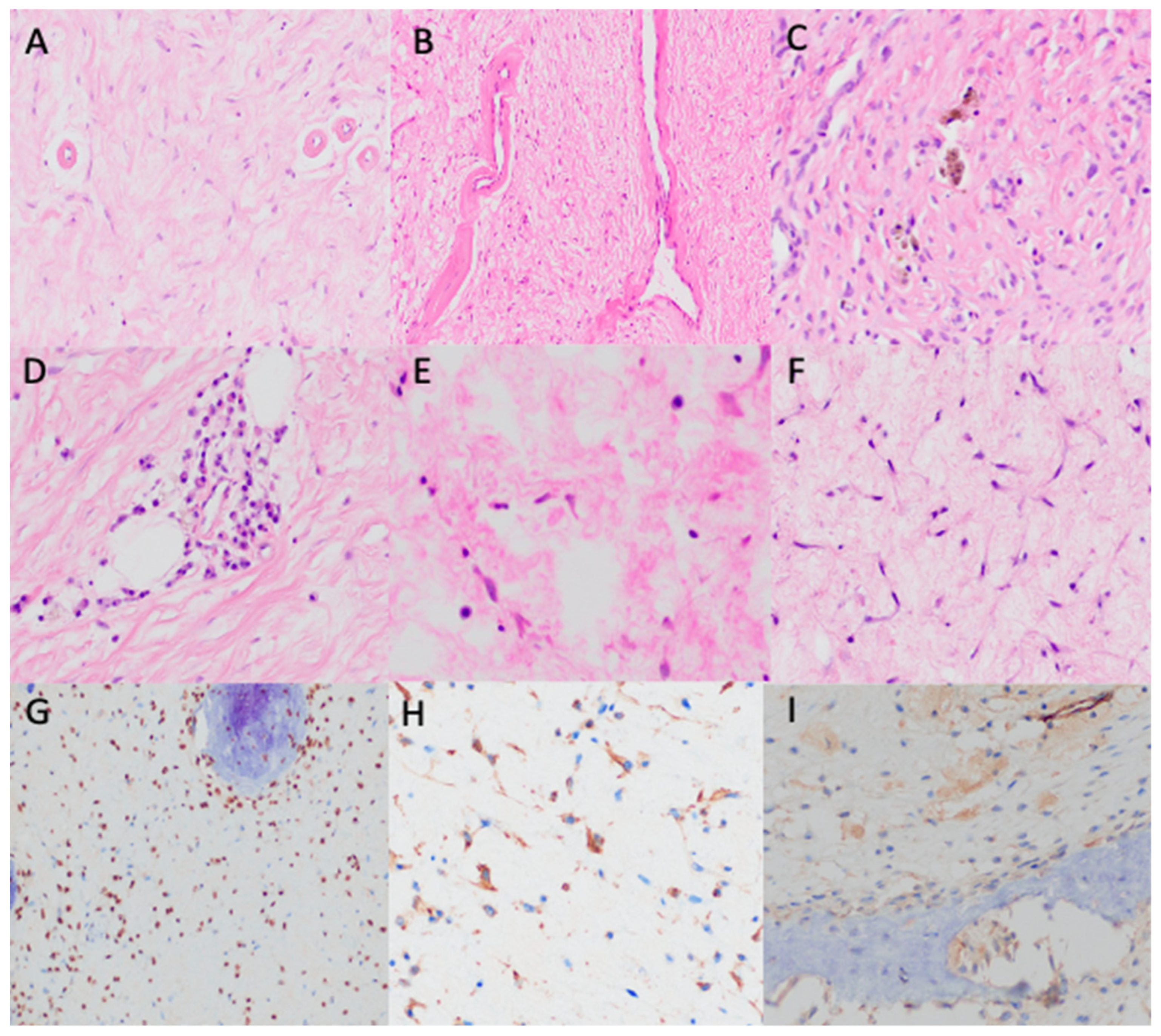
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deel, C.; Hassell, L. Liposclerosing Myxofibrous Tumor: A Review. Arch. Pathol. Lab. Med. 2016, 140, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Kransdorf, M.J.; Murphey, M.D.; Sweet, D.E. Liposclerosing myxofibrous tumor: A radiologic-pathologic-distinct fibro-osseous lesion of bone with a marked predilection for the intertrochanteric region of the femur. Radiology 1999, 212, 693–698. [Google Scholar] [CrossRef]
- Campbell, K.; Wodajo, F. Two-step Malignant Transformation of a Liposclerosing Myxofibrous Tumor of Bone. Clin. Orthop. 2008, 466, 2873–2877. [Google Scholar] [CrossRef] [PubMed]
- Bahk, W.J.; Seo, K.J. Malignant transformation of liposclerosing myxofibrous tumour. Pathology 2021, 53, 660–663. [Google Scholar] [CrossRef]
- Illac, C.; Delisle, M.B.; Bonnevialle, P.; Chiavassa-Gandois, H.; de Pinieux, G.; Gomez-Brouchet, A. Telangiectatic osteosarcoma secondary to a liposclerosing myxofibrous tumor: A case report. Ann. Pathol. 2012, 32, 259–262. [Google Scholar] [CrossRef]
- Heim-Hall, J.M.; Williams, R.P. Liposclerosing myxofibrous tumour: A traumatized variant of fibrous dysplasia? Report of four cases and review of the literature. Histopathology 2004, 45, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Dattilo, J.; McCarthy, E.F. Liposclerosing myxofibrous tumor (LSMFT), a study of 33 patients: Should it be a distinct entity? Iowa Orthop. J. 2012, 32, 35–39. [Google Scholar]
- Siegal, G.; Bloem, J.; Cates, J.; Hameed, M.; WHO Classification of Tumours Editorial Board. Fibrous Dysplasia. In Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Geneva, Switzerland, 2020; pp. 472–474. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, B.D.; Sweet, D.E. Bone. In the Pathology of Incipient Neoplasia; Saunders: Philadelphia, PA, USA, 1986; pp. 381–423. [Google Scholar]
- Gilkey, F.W. Liposclerosing myxofibrous tumor of bone. Hum. Pathol. 1993, 24, 1264. [Google Scholar] [CrossRef] [PubMed]
- Matsuba, A.; Ogose, A.; Tokunaga, K.; Kawashima, H.; Hotta, T.; Urakawa, S.; Umezu, H.; Higuchi, T.; Endo, N. Activating Gs alpha mutation at the Arg201 codon in liposclerosing myxofibrous tumor. Hum. Pathol. 2003, 34, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, H.M.; Al-Nakshabandi, N.A.; Saliken, J.; Munk, P.L.; Nielsen, T.O.; Masri, B.; O’connell, J.X. Liposclerosing myxofibrous tumour. Eur. J. Radiol. Extra 2005, 55, 83–87. [Google Scholar] [CrossRef]
- Corsi, A.; De Maio, F.; Ippolito, E.; Cherman, N.; Robey, P.G.; Riminucci, M.; Bianco, P. Monostotic fibrous dysplasia of the proximal femur and liposclerosing myxofibrous tumor: Which one is which? J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2006, 21, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.C.K.; Rao, C.G. Polymorphic fibro-osseous lesion of the bone: A case report with review of literature. Indian. J. Pathol. Microbiol. 2008, 51, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Nieto, A.; Pérez-Andrés, R.; Lorenzo, J.C.; Vilanova, J.C. Diagnostic imaging of liposclerosing myxofibrous tumor of bone. Radiologia 2010, 52, 251–254. [Google Scholar] [CrossRef] [PubMed]
- González Ortega, F.J.; Peñas García, J.; Ramírez Garrido, F. Tumor fibromixoide lipoesclerosante. Rev. Argent. Radiol. 2015, 79, 222–223. [Google Scholar] [CrossRef]
- Técualt-Gómez, R.; Atencio-Chan, A.; Cario-Méndez, A.G.; Amaya-Zepeda, R.A.; Balderas-Martinez, J.; González-Valladares, J.R. Bone liposclerosing myxofibrous tumor. Case presentation and literature review. Acta Ortop. Mex. 2015, 29, 191–195. [Google Scholar]
- Kim, J.; Chen, W.; Resnik, C.; Dilsizian, V.; Chen, Q.; Kimball, A.S. FDG uptake in liposclerosing myxofibrous tumor causes upstaging of Hodgkin lymphoma. Clin. Nucl. Med. 2015, 40, 325–327. [Google Scholar] [CrossRef]
- Regado, E.R.; Garcia, P.B.L.; Caruso, A.C.; de Almeida, A.L.B.; Aymoré, I.L.; Meohas, W.; Aguiar, D.P. Liposclerosing myxofibrous tumor: A series of 9 cases and review of the literature. J. Orthop. 2016, 13, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Kampen, W.U.; Antunovic, L.; Luebke, A.M.; Sauter, G.; Strobel, K.; Paycha, F. Bone SPECT/CT imaging of a liposclerosing myxofibroid tumor in an unexpected localization. Eur. J. Hybrid. Imaging 2018, 2, 29. [Google Scholar] [CrossRef]
- Ploof, J.; Shaikh, H.; Melli, J.; Jour, G.; Turtz, A. Liposclerosing Myxofibrous Tumor of the Cranial Vault: A Case Report. Neurosurgery 2019, 84, E207–E210. [Google Scholar] [CrossRef]
- Pai, S.N.; Kumar, M.M. Liposclerosing myxofibrous tumour. BMJ Case Rep. 2021, 14, e245487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, D.; Yu, W.; Wang, C. Liposclerosing myxofibrous tumor of the distal femur: A case report. Front. Surg. 2022, 9, 1009975. [Google Scholar] [CrossRef] [PubMed]
- Beytemür, O. Liposclerosing myxofibrous tumor: A rare tumor of proximal femur. Jt. Dis. Relat. Surg. 2017, 28, 210–213. [Google Scholar] [CrossRef]
- Choi, J.W.; Lee, Y.S.; Lee, J.H.; Kim, H.K.; Yeom, B.W.; Choi, J.S.; Lim, H.C.; Kim, C.H. Liposclerosing Myxofibrous Tumor in Tibia—A Case Report and Review of the Literature. Korean J. Pathol. 2005, 39, 207–210. [Google Scholar]
- Teruel-González, V.; Vicente-Zuloaga, M.; Oncalada-Calderón, E. Liposclerosing myxofi brous hip tumour. A case. Rev. Esp. Cir. Ortop. Traumatol. 2010, 54, 120–125. [Google Scholar]
- Ragsdale, B.D. Polymorphic fibro-osseous lesions of bone: An almost site-specific diagnostic problem of the proximal femur. Hum. Pathol. 1993, 24, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Unni, K.K.; Mertens, F.; WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, 3rd ed.; IARC Press: Geneva, Switzerland, 2002. [Google Scholar]
- Rosenberg, A.E.; Cleton-Jansen, A.M.; de Pinieux, G.; Deyrup, A.T.; Hauben, E.; Squire, J. Osteosarcoma. In WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, 4th ed.; IARC Press: Geneva, Switzerland, 2013. [Google Scholar]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials. Cancers 2023, 15, 429. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agaram, N.P.; Bredella, M.A.; WHO Classification of Tumours Editorial Board. Aneurysmal bone cyst. In Soft Tissue and Bone Tumours, 5th ed.; Agency for Research on Cancer: Lyon, France, 2020; pp. 437–439. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Case | Age, Sex | Location | Symptoms | Radiology | Histology | Molecular Tests | Treatment | Evolution | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Side | Bone | Site | X-Ray/CT | MRI/PET/SPECT | Stroma | Cells | Bone | Adipose | Xantomized Cells | Cystic Spaces | Vessels | Inflammation | Necrosis | Other | Malignant Transformation | ||||||||
| Micro | Macro | ||||||||||||||||||||||
| 1 | M, 26 | R | Femur | P | IT | Pathological fracture | Ly, Sc rim | INA | 95% Fibromixoid | Stellated, spindled No atipia | 3% Psamomatoid | No | No | 2% | No | Small, hyalinized | No | No | - | No | Not performed | Curettage and bone grafting | 312 months, NED |
| 2 | F, 24 | R | Femur | P | IT | ID | INA | INA | 75% Fibromixoid | Stellated, spindled No atipia | 4% Psamomatoid | 15% Peripheral | 1% | 5% | No | Small, hyalinized | Mast cells | No | - | No | Not performed | Curettage and bone grafting | 215 months, NED |
| 3 | M, 61 | R | Femur | P | IT | ID (Hodgkin lymphoma extension study) | INA | INA | 20% Fibromixoid | Stellated, spindled No atipia | 40% Gross trabeculae, Pagatoid | 40% Intermixed | No | No | No | Small, hyalinized | Mast cells, lymphocytes | No | - | No | Not performed | Curettage and bone grafting | 192 months, NED Death (multi-organ failure) |
| 4 | M, 19 | L | Femur | D | - | Pain | Ly, cystic, internal trabeculae | Ly, cystic, internal trabeculae T2WI: HI Fat areas | 80% Hyalinized | Stellated, spindled No atipia | 3% Psamomatoid | 10% Intermixed | 6% | No | 1% | Small, hyalinized | No | No | - | No | Not performed | Curettage and bone grafting, nail-plate | 281 months, NED |
| 5 | M, 28 | L | Femur | P | IT | ID | Ly, Sc rim | Ly, Sc rim T1WI: isointense T2WI: HI | 95% Fibromixoid | Stellated, spindled No atipia | 2% Trabeculae | No | 3% | No | No | Small, hyalinized | Mast cells | No | - | No | Not performed | Curettage and bone grafting, nail-plate | 145 months, NED |
| 6 External case | F, 48 | R | Femur | P | IT | Pain | Ly, cystic, internal trabeculae | INA | 75% Fibromixoid | Stellated, spindled No atipia | 10% Trabeculae | 2% Intermixed | No | No | 3% | Small, hyalinized | No | 10% Ischemic | ABC –like areas | No | Not performed | Curettage and bone grafting | 303 months, NED |
| 7 | F, 20 | L | Femur | P | IT | Pain Previous local trauma | INA | INA | 95% Fibrous | Stellated, spindled No atipia | 2% Psamomatoid | No | 1% | 3% | No | Small, hyalinized | No | No | - | No | Not performed | Curettage and bone grafting, nail-plate | 134 months, NED |
| 8 | M, 68 | R | Femur | P | IT | ID | Ly, Sc rim | Sc rim, internal septae PET: no uptake | 70% Fibromixoid | Stellated, spindled No atipia | 30% * Trabeculae * CNB | No | No | No | No | Not seen | Plasma cells | No | - | No | Not performed | Curettage and bone grafting | 167 months, NED |
| 9 | M, 19 | R | Femur | P | IT | Pain | WD, Ly, Sc rim | Bone scintigraphy and PET: peripheral uptake | 77% Mixoid > fibro | Stellated, spindled No atipia | 3% Trabeculae | No | No | 15% | 5% | Small, hyalinized | Lymphocytes, hemosiderophages | 1% Ischemic | - | No | NGS: TP53 mutation (c.520A > T, p.Arg174Trp) | Curettage and bone grafting, nail plate | 38 months, NED |
| 10 | F, 50 | L | Femur | P | IT | ID (ovarian tumor extension study) | WD, Ly, Sc rim | T1WI: hi T2WI: HI SPECT: moderate homogeneous uptake | 50% Fibromixoid | Stellated, spindled No atipia | 50% * Trabeculae * CNB | No | No | No | No | Not seen | No | No | - | No | Not performed | Expectant management | 13 months, NED Death (ovarian tumor) |
| 11 | F, 52 | L | Femur | P | IT | Pain | WD, Ly, Sc rim | T1–WI: hi T2–WI: HI Fat areas | 90% Fibromixoid, dense collagen bands | Stellated, spindled No atipia | 5% Psamomatoid | No | 2% | No | 3% | Small, hyalinized | Lymphocytes | No | - | No | NGS: GNAS mutation (c.2530C > T, p.Arg844Cys ) | Curettage and bone grafting | 41 months, NED |
| 12 External case | F, 72 | L | Femur | P | IT | Pain | WD, Ly, Sc rim | INA | 94% Fibromyxoid | Stellated, spindled No atipia | 5% Psamomatoid | No | No | 1% | No | Not seen | No | No | - | No | Not performed | Curettage and bone grafting | 240 months, NED Natural death |
| 13 | M, 38 | R | Femur | P | IT | Pathological fracture | INA | T1WI: hi T2WI: HI Fat areas | 25% Fibromyxoid | Stellated, spindled No atipia | 30% Pagetoid | 10% Intermixed | 5% | No | No | Small, hyalinized | No | No | Bone infarct (30%) | No | Not performed | Curettage and bone grafting | 257 months, NED |
| 14 External case | F, 26 | L | Femur | P | IT | ID | INA | INA | 72% Fibromyxoid | Stellated, spindled No atipia | 15% Ttrabeculae, pagatoid, psamomatoid | 1% Intermixed | 4% | 8% | No | Small, hyalinized | Perivascular plasma cells and lymphocytes | No | - | No | Not performed | Curettage and bone grafting, nail plate | 129 months, NED |
| 15 | M, 18 | R | Femur | P | IT | Pain | INA | INA | 60% Mixoid > fibro | Stellated, spindled No atipia | 30% Trabeculae | 10% Intermixed | No | No | No | Big, hyalinized | Hemosiderophages | No | - | No | Not performed | Curettage and bone grafting | 131 months, NED |
| Authors, Year | Nº of Cases | Age, Sex | Location | Symptoms | Radiology | Histology | Treatment | Evolution | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Side | Bone | Site | X-Ray/CT | MRI/PET/SPECT | Stroma/Cells | Bone | Adipose | Xantom. Cells | Cystic Spaces | Other | Malignant Transformation | |||||||
| Ragsdale et al., 1993 [28] | 95 | 21–67 (mean 43) | INA | Femur (87) Tibia (2) Humerus (4) Cranial vault (2) | Femur: P (85), D (2) | INA | ID, pain, pathological fracture | WD, Ly, Sc rim, increase uptake | INA | INA | WB, LB Pagetoid | Yes | Yes | INA | Cartilage areas (rare) | Yes (15) | INA | INA |
| Gilkey et al., 1993 [11] | 40 | INA | INA | INA | INA | INA | INA | INA | INA | INA | Yes (1) | INA | INA | |||||
| Krandsorf et al., 1999 [2] | 39 | 15–69 (mean 42) M (21), F (18) | INA | Femur (33) Iliac bone (3), humerus (2) Rib (1) | Femur: P (30), shaft (3) | IT (30) | Pain (14), ID (12), pathological fracture (3) | Ly, WD, geographic, variable Sc areas, variable mineralized matrix | Well-defined, variable-thickness peripheral rim, T1WI: isointense, homogeneous, T2WI: HI, heterogeneous, STIR: HI | INA | Yes (4): osteosarcoma (2), malignant fibrous hystiocitoma (1), low grade malignant change (1) | INA | INA | |||||
| Heim-Hall et al., 2003 [6] | 4 | 14, M | R | Femur | P | ST | Pathological fracture | Ly, ground-glass opacity, scalloped endosteum | INA | Fibromyxoid | Trabecular (FD-like) | Fat necrosis | Yes | No | - | No | Curettage | INA |
| 74, F | L | Femur | P | IT, ST | Pain | Cystic lesion, Sc margins | INA | Fibromyxoid | Pagetoid Bone necrosis | Yes | Yes | No | - | No | Curettage and grafting | INA | ||
| 37, F | L | Femur | - | - | Pain | WD, lucent, thick bony borders | INA | Fibromyxoid | Trabecular (FD-like) | Yes | Yes | No | - | No | Curettage | INA | ||
| 37, F | R | Femur | - | - | Pathological fracture | Ly, ground-glass opacity, scalloped endosteum | INA | Fibromyxoid | Trabecular (FD-like) | Fat necrosis | No | No | ABC component | No | Curettage and grafting | INA | ||
| Matsuba et al., 2003 [12] | 2 | 75, M | R | Femur | P | IT | Pain | WD, Ly, Sc rim | T2WI: hi and HI | Fibromyxoid | Curvilinear, trabecular (FD-like) | Yes | Yes | No | GNAS mutation | No | Curettage and grafting | 16 months, NED |
| 59, M | INA | Iliac bone | - | - | ID | WD, Ly, Sc rim | T2WI: hi and HI | Fibromyxoid | Curvilinear, trabecular (FD-like) | Yes | Yes | No | GNAS mutation | No | Curettage and grafting | 24 months, NED | ||
| O’Dwyer et al., 2005 [13] | 1 | 51, M | L | Femur | P | IT | Pain | WD, Ly, Sc rim | T1WI: hi T2WI: HI STIR: HI | Fibromyxoid | WB Trabecular (FD-like), pagetoid, spicules | Yes | Yes | No | Dystrophic calcification | No | INA | INA |
| Choi et al., 2005 [26] | 1 | 61, M | L | Tibia | P | Diaphysis | ID | WD, Ly, Sc rim | T1WI: hi T2WI: HI Heterogeneous enhancement | Fibromyxoid | WB Trabecular (FD-like), pagetoid | Yes | Yes | No | - | No | Curettage | INA |
| Corsi et al., 2006 [14] | 2 | 33, F | L | Femur | P | Neck | Multiple bone lesions: monostotic FD | Ground-glass lesion, increased uptake | INA | INA | Trabecular (FD-like) | Yes | Yes | Yes | No GNAS mutation detected | No | Curettage | INA |
| 54, F | L | Femur | P | IT | ID | Lucent lesion with scattered densities and a Sc rim | INA | Fibromyxoid | Trabecular woven bone | No | Yes | No | GNAS mutation | No | Curettage and grafting | INA | ||
| Rao et al., 2008 [15] | 1 | 35, F | L | Femur | P | IT | Pain, prior local trauma | WD, Ly, ground-glass opacity, scalloped endosteum | INA | Fibromyxoid | Spicules | No | Yes | No | Scattered lymphocytes | No | INA | INA |
| Campbell et al., 2008 [3] | 1 | 42, F | L | Femur | P | IT | Pain and pathological fracture | WD, Ly, Sc rim | INA | Fibromyxoid | INA | Yes | Yes | No | - | Yes, OS with giant cells | Curettage and cemented fixation | 31 months, death (lung metastases) |
| Teruel-González et al., 2008 [27] | 1 | 43, M | L | Femur | P | IT | ID | WD, Ly, internal Ca | T1WI: hi T2WI: HI | Fibromyxoid | WB Pagetoid, trabecular (FD-like) | Yes | Yes | No | - | No | Curettage and grafting | NED |
| Nieto et al., 2009 [16] | 2 | 50, M | R | Femur | P | IT | ID (femoral chondrosarcoma extension study) | WD, Sc rim | PET: increased activity | Fibromyxoid | Yes | No | Yes | No | - | No | INA | INA |
| 57, M | R | Femur | P | IT | Pain | WD, Ly, Sc rim, internal Ca | T1WI: hi T2-WI: HI | INA | INA | INA | ||||||||
| Dattilo et al., 2012 [7] | 33 | 19–80 (mean 46) M (20), F (13) | INA | Femur | P | IT | ID (21) Pain (10) Pathological fracture (2) | WD, geographic lucencies, Sc rim Ground-glass areas (15) | T2WI: heterogeneous | Fibromyxoid | Reactive bone, woven bone | Yes (3) | No | No | - | No | Curettage and grafting (13) | INA |
| Illac et al., 2012 [5] | 1 | 84 | L | Femur | P | IT | Pain Multiple bone lesions | WD, Ly, hipodense | T2WI: HI Heterogeneous enhancement | INA | INA | INA | Yes | INA | - | Yes, TG OS | INA | INA |
| González et al., 2014 [17] | 1 | 25, M | L | Femur | P | IT | Pain | WD, Ly, Sc rim, internal Ca | INA | Fibromixoid | Trabecular (FD-like) | Yes | Yes | No | - | No | INA | INA |
| Técualt-Gómez et al., 2015 [18] | 1 | 80, M | R | Femur | P | IT | Pain | WD, Ly, Sc rim, internal Ca | T1WI: hi T2WI: HI | Fibromixoid | Trabecular (FD-like) Bone necrosis | Fat Necrosis | No | No | Dystrophic calcification | No | Curettage and PMMA | INA |
| Kim et al., 2015 [19] | 1 | 22, F | R | Femur | P | INA | ID (Hodgkin lymphoma extension study) | WD, Ly, Sc rim | PET: intense uptake | Fibromixoid | Trabecular | No | No | No | - | No | INA | INA |
| Regado et al., 2016 [20] | 9 | 50, F | R | Femur | P | IT | Pain and limping Local edema (2) Local trauma (2) Pathological fracture (3) | WD, Ly, Sc rim | INA | Fibromixoid Stellate, fusiform, bipolar and oval cells | Bone necrosis | Yes | Yes | Yes | - | Yes (1) Increased cellularity and atypical nuclei | Curettage and grafting | 14 months, NED |
| 53, M | R | Tibia | D | - | WD, Ly, Sc rim, cortical disruption | Transtibial amputation | 97 months, NED | |||||||||||
| 19, M | R | Femur | D | - | WD, Ly, Sc rim | Curettage and grafting | 76 months, NED | |||||||||||
| 45, F | R | Femur | P | IT | WD, Ly, Sc rim | Curettage and grafting | 66 months, NED | |||||||||||
| 42, F | L | Femur | P | IT | WD, Sc | Curettage and grafting | 66 months, NED | |||||||||||
| 50, F | L | Iliac bone | - | - | WD, Ly, Sc rim | No treatment | 40 months, NED | |||||||||||
| 39, F | R | Femur | P | IT | WD, Ly, Sc rim | Curettage and grafting | 29 months, NED | |||||||||||
| 30, F | R | Femur | P | IT | WD, Ly, Sc rim | Curettage and grafting | 28 months, NED | |||||||||||
| 27, F | L | Femur | P | IT | WD, Ly, Sc rim | Curettage and grafting | 14 months, NED | |||||||||||
| Beytemür et al., 2017 [25] | 1 | 67, M | R | Femur | P | IT | Pain | WD, Sc rim, internal septations | T1WI: hi T2WI: HI Heterogeneous enhancement PET: increased activity | Fibromixoid | Trabecular (FD-like) | No | Yes | No | - | No | Curettage | 16 months, NED |
| Kampen et al., 2018 [21] | 1 | 69, M | L | Iliac bone | - | - | Pain | WD, Sc rim | T2WI: HI Slight Enhancement SPECT/CT: EP: no abnormal hyperperfusion DP: increased activity | Fibrous | WB, LB Curvilinear, pagetoid | No | Yes | No | - | No | INA | INA |
| Ploof et al., 2019 [22] | 1 | 30, F | L | Cranial vault | - | - | Slow growing painless mass | WD, expansile, scalloped endosteum, scattered peripheral Ca | Heterogeneous enhancement | Fibromixoid | WB Curvilinear | Yes | No | Yes | Dystrophic calcification | No | Complete excision | 8 months, NED |
| Bahk et al., 2020 [4] | 1 | 75, F | R | Femur | P | IT | Multiple bone lesions: polyostotic FD, AVN (left femur) | Lobular, ground-glass opacity | Heterogeneous T1WI: hi T2WI: HI | Fibrous | WB Pagetoid, spicules | Yes | No | No | - | Yes, high grade sarcoma (FS) | Bipolar hemiarthroplasty with cementation | 5 months, death (multiple metastases) |
| Pai et al., 2021 [23] | 1 | 63, M | L | Femur | P | IT, ST | ID | WD, Ly, Sc rim, internal septations | Heterogeneous T1WI: hi T2WI: HI | INA | No | Conservative management | 6 months, NED | |||||
| Zhang et al., 2022 [24] | 1 | 55, F | L | Femur | D | - | Pain | Patchy high-density shadow | Heterogeneous T1WI: hi T2WI: HI | Collagenoid, myxoid degeneration | Yes | Fat necrosis | No | Yes | - | No | Curettage and bone grafting | 6 months, NED |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pena-Burgos, E.M.; Serra del Carpio, G.; Tapia-Viñe, M.; Suárez-González, J.; Buño, I.; Ortiz-Cruz, E.; Pozo-Kreilinger, J.J. Liposclerosing Myxofibrous Tumor: A Separated Clinical Entity? Diagnostics 2025, 15, 536. https://doi.org/10.3390/diagnostics15050536
Pena-Burgos EM, Serra del Carpio G, Tapia-Viñe M, Suárez-González J, Buño I, Ortiz-Cruz E, Pozo-Kreilinger JJ. Liposclerosing Myxofibrous Tumor: A Separated Clinical Entity? Diagnostics. 2025; 15(5):536. https://doi.org/10.3390/diagnostics15050536
Chicago/Turabian StylePena-Burgos, Eva Manuela, Gabriela Serra del Carpio, Mar Tapia-Viñe, Julia Suárez-González, Ismael Buño, Eduardo Ortiz-Cruz, and Jose Juan Pozo-Kreilinger. 2025. "Liposclerosing Myxofibrous Tumor: A Separated Clinical Entity?" Diagnostics 15, no. 5: 536. https://doi.org/10.3390/diagnostics15050536
APA StylePena-Burgos, E. M., Serra del Carpio, G., Tapia-Viñe, M., Suárez-González, J., Buño, I., Ortiz-Cruz, E., & Pozo-Kreilinger, J. J. (2025). Liposclerosing Myxofibrous Tumor: A Separated Clinical Entity? Diagnostics, 15(5), 536. https://doi.org/10.3390/diagnostics15050536