
A Robust and Comprehensive Study of the Molecular and Genetic Basis of Neurodevelopmental Delay in a Sample of 3244 Patients, Evaluated by Exome Analysis in a Latin Population
 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Sampling
2.2. Review of Medical Records
2.3. Sampling, Sequencing, Bioinformatics Processing, and Variant Filtering
2.4. Classification and Ontology Analysis of Associated Genes
2.5. Statistical Analysis
2.6. Ethical Considerations
3. Results
3.1. Diagnostic Yield
3.2. Characterization of Novel Variants
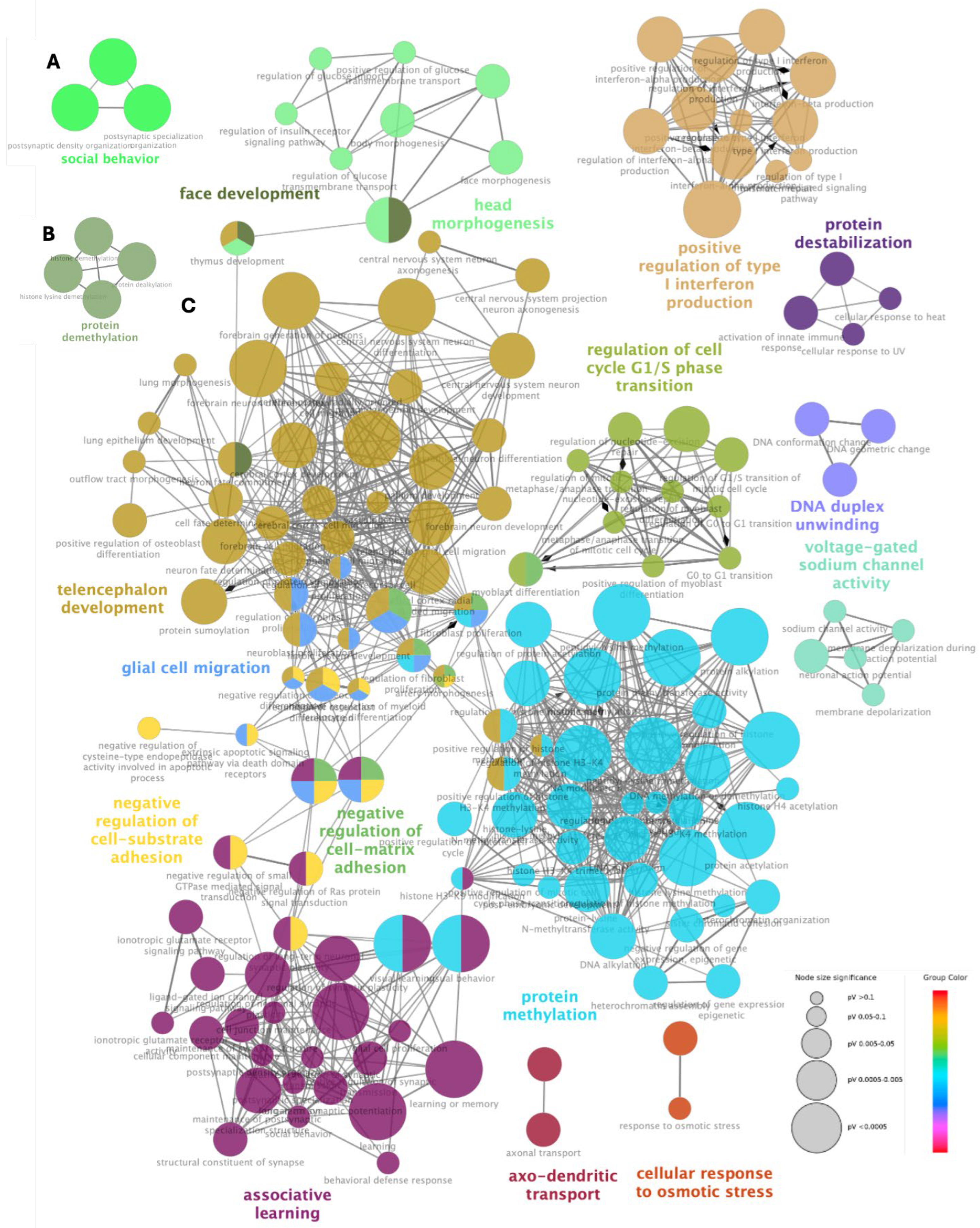
3.3. Gene Ontology Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gidziela, A.; Ahmadzadeh, Y.I.; Michelini, G.; Allegrini, A.G.; Agnew-Blais, J.; Lau, L.Y.; Duret, M.; Procopio, F.; Daly, E.; Ronald, A.; et al. A meta-analysis of genetic effects associated with neurodevelopmental disorders and co-occurring conditions. Nat. Hum. Behav. 2023, 7, 642–656. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders and the NDD Exome Scoping Review Work Group. Genet. Med. 2019, 21, 2413–2421. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.F.; Chahrour, M.H.; Walsh, C.A. The diverse genetic landscape of neurodevelopmental disorders. Annu. Rev. Genom. Hum. Genet. 2014, 15, 195–213. [Google Scholar] [CrossRef]
- Wayhelova, M.; Vallova, V.; Broz, P.; Mikulasova, A.; Smetana, J.; Dynkova Filkova, H.; Machackova, D.; Handzusova, K.; Gaillyova, R.; Kuglik, P. Exome sequencing improves the molecular diagnostics of paediatric unexplained neurodevelopmental disorders. Orphanet J. Rare Dis. 2024, 19, 41. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; de Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Griffin, A.; Mahesh, A.; Tiwari, V.K. Disruption of the gene regulatory programme in neurodevelopmental disorders. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194860. [Google Scholar] [CrossRef] [PubMed]
- Ballesta-Martínez, M.J.; Pérez-Fernández, V.; López-González, V.; Sánchez-Soler, M.J.; Serrano-Antón, A.T.; Rodríguez-Peña, L.I.; Barreda-Sánchez, M.; Armengol-Dulcet, L.; Guillén-Navarro, E. Validation of clinical exome sequencing in the diagnostic procedure of patients with intellectual disability in clinical practice. Orphanet J. Rare Dis. 2023, 18, 201. [Google Scholar] [CrossRef]
- Martinez-Granero, F.; Blanco-Kelly, F.; Sanchez-Jimeno, C.; Avila-Fernandez, A.; Arteche, A.; Bustamante-Aragones, A.; Rodilla, C.; Rodríguez-Pinilla, E.; Riveiro-Alvarez, R.; Tahsin-Swafiri, S.; et al. Comparison of the diagnostic yield of aCGH and genome-wide sequencing across different neurodevelopmental disorders. NPJ Genom. Med. 2021, 6, 25. [Google Scholar] [CrossRef]
- Monroe, G.R.; Frederix, G.W.; Savelberg, S.; De Vries, T.I.; Duran, K.J.; Van Der Smagt, J.J.; Terhal, P.A.; Van Hasselt, P.M.; Kroes, H.Y.; Verhoeven-Duif, N.M.; et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet. Med. 2016, 18, 949–956. [Google Scholar] [CrossRef]
- Vrijenhoek, T.; Middelburg, E.M.; Monroe, G.R.; van Gassen, K.L.; Geenen, J.W.; Hövels, A.M.; Knoers, N.V.; van Amstel, H.K.; Frederix, G.W. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. Eur. J. Hum. Genet. 2018, 26, 1566–1571. [Google Scholar] [CrossRef]
- Savatt, J.M.; Myers, S.M. Genetic testing in neurodevelopmental disorders. Front. Pediatr. 2021, 9, 526779. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; Obiajulu, J.U.; Wright, J.R.; Murali, S.C.; Xu, S.X.; et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 2022, 54, 1305–1319. [Google Scholar] [CrossRef] [PubMed]
- Servetti, M.; Pisciotta, L.; Tassano, E.; Cerminara, M.; Nobili, L.; Boeri, S.; Rosti, G.; Lerone, M.; Divizia, M.T.; Ronchetto, P.; et al. Neurodevelopmental disorders in patients with complex phenotypes and potential complex genetic basis involving non-coding genes, and double CNVs. Front. Genet. 2021, 12, 732002. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Zhang, X.; Lei, Q.; Chen, P.; Hu, H.; Shen, S.; Liu, J.; Ye, S. Case report: Genetic analysis of a novel frameshift mutation in FMR1 gene in a Chinese family. Front. Genet. 2023, 14, 1228682. [Google Scholar] [CrossRef]
- Leite, A.J.; Pinto, I.P.; Leijsten, N.; Ruiterkamp-Versteeg, M.; Pfundt, R.; de Leeuw, N.; da Cruz, A.D.; Minasi, L.B. Diagnostic yield of patients with undiagnosed intellectual disability, global developmental delay and multiples congenital anomalies using karyotype, microarray analysis, whole exome sequencing from Central Brazil. PLoS ONE 2022, 17, e0266493. [Google Scholar] [CrossRef]
- Bruno, L.P.; Doddato, G.; Valentino, F.; Baldassarri, M.; Tita, R.; Fallerini, C.; Bruttini, M.; Lo Rizzo, C.; Mencarelli, M.A.; Mari, F.; et al. New candidates for autism/intellectual disability identified by whole-exome sequencing. Int. J. Mol. Sci. 2021, 22, 13439. [Google Scholar] [CrossRef]
- Wilfert, A.B.; Sulovari, A.; Turner, T.N.; Coe, B.P.; Eichler, E.E. Recurrent de novo mutations in neurodevelopmental disorders: Properties and clinical implications. Genome Med. 2017, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Vanderhaeghen, P.; Polleux, F. Developmental mechanisms underlying the evolution of human cortical circuits. Nat. Rev. Neurosci. 2023, 24, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Swanberg, S.E.; Nagarajan, R.P.; Peddada, S.; Yasui, D.H.; LaSalle, J.M. Reciprocal co-regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Hum. Mol. Genet. 2009, 18, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Schlotthauer, I.; Kizner, V.; Macartney, T.; Dorner-Ciossek, C.; Gillardon, F. Loss-of-function Mutations of CUL3, a High Confidence Gene for Psychiatric Disorders, Lead to Aberrant Neurodevelopment In Human Induced Pluripotent Stem Cells. Neuroscience 2020, 448, 234–254. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Yang, J.; Wu, S.; Ye, T.; Zhuang, W.; Wang, W.; Tan, T. Current trends of high-risk gene Cul3 in neurodevelopmental disorders. Front. Psychiatry 2023, 14, 1215110. [Google Scholar] [CrossRef]
- Uchino, S.; Waga, C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev. 2013, 35, 106–110. [Google Scholar] [CrossRef]
- Heiman, P.; Drewes, S.; Ghaloul-Gonzalez, L. A familial case of CAMK2B mutation with variable expressivity. SAGE Open Med. Case Rep. 2021, 9, 2050313X21990982. [Google Scholar] [CrossRef]
- Pande, S.; Majethia, P.; Nair, K.; Rao, L.P.; Mascarenhas, S.; Kaur, N.; do Rosario, M.C.; Neethukrishna, K.; Chaurasia, A.; Hunakunti, B.; et al. De novo variants underlying monogenic syndromes with intellectual disability in a neurodevelopmental cohort from India. Eur. J. Hum. Genet. 2024, 32, 1291–1298. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Phenotype | Gene |
|---|---|
| Developmental delay | ACTL6B, AHDC1, ANKRD11, ARID2, ATRX, BCL11A, BCL11B, BRAF, CHD3, CREBBP, CTNNB1, CUL3, DDX3X, DNM1, DNMT3A, DPYD, DYNC1H1, FBXO11, FLNB, FOXG1, GLUD2, GNB1, GRIA2, GRIN1, H1-4, IFIH1, INTS1, IVD, JAG1, KCNT2, KDM6B, KIAA1109, KIF1A, KMT2A, KMT2C, LARP7, LARS2, LMAN2L, LZTR1, MECP2, MN1, MSTO1, NACC1, NALCN, NARS, NEXMIF, NF1, NFIB, PHF8, PIK3R1, PPM1D, PTPN11, PURA, QRICH1, RAF1, RAI1, RBMX, RPS6KA3, SATB2, SCN1A, SCN2A, SCN8A, SETD2, SETD5, SHANK2, SHANK3, SIX3, SLC13A5, SPEN, STAG2, SYNGAP1, TAOK1, TBK1, TBR1, TCF20, TCF4, TOE1, TREX1, TRIP12, TSC2, TSPAN7, TUBB, UBTF, USP7, WAC, ZMIZ1, AP1S2, ARSA, CAPN3, CDC42, COL6A2, COL6A3, CTBP1, DCX, DHTKD1, EP300, FBN2, GCDH, PMM2, POLR3B, PUF60, RTN2, RYR1, SPG11, SPG7, SPTBN2, TNPO3, TRPV4, TRIO, RNASEH2B, RFX7, KMT2E, ATP7B, ARID1B, INPP5E, KDM3B, CSNK2A1 |
| Developmental delay and epilepsy | ABCC8, AFF3, ANKRD17, CDKL5, CHD2, GNAO1, GRIN2B, HNRNPU, KAT6A, KCNB1, KCNMA1, PTCH1, SETD1A, SLC2A1, STXBP1, MN1 |
| Autism spectrum disorder | CACNA1E, CAMK2A, CHD8, CPT2, CREBBP, CSF1R, CUL3, DNM1, EBF3, KDM4B, PTEN, SHANK2, SHANK3 |
| Cognitive impairment | KDM4B, KDM5C, KDM6A, PAX8, NAA15, KDM3B, SHANK3 |
| Gene | DNA | Protein | Variant Type | ACMG Classification | Transcripts | OMIM Code |
|---|---|---|---|---|---|---|
| ACTL6B # | c.521_522insA | p.Thr175Hisfs*7 | Frameshift | LP (PVS1, PM2) | NM_016188.4 | 612458 |
| ACTL6B * | c.991G>A | p.Gly331Ser | Missense | LP (PS2, PM2, PP2) | NM_016188.5 | 612458 |
| AHDC1 | c.4294del | p.Ala1432Profs*13 | Frameshift | LP (PVS1, PM2) | NM_001371928.1 | 615790 |
| ANKRD11 | c.741C>A | p.Tyr247* | Nonsense | LP (PVS1, PM2) | NM_013275.6 | 611192 |
| ANKRD11 * | c.7124_7152del | p.Glu2375Alafs*147 | Frameshift | P (PVS1, PS2, PM2) | NM_013275.5 | 611192 |
| ANKRD17 * | c.4453_4457del | p.Lys1485Glufs*17 | Frameshift | P (PVS1, PS2, PM2) | NM_032217.4 | 615929 |
| AP1S2 # | c.180-1G>C | Splicing site | LP (PVS1, PM2) | NM_001272071.2 | 300629 | |
| BCL11B * | c.2345dup | p.Gly785Argfs*100 | Frameshift | P (PVS1, PS2, PM2) | NM_138576.3 | 606558 |
| BRAF * | c.2030A>G | p.Asp677Gly | Missense | P (PS2, PM1, PM2, PP2, PP3) | NM_004333.6 | 164757 |
| CACNA1E * | c.5365-2A>G | - | Splicing site | LP (PS2, PM2) | NM_001205293.1 | 601013 |
| CHD2 * | c.2822A>T | p.Gln941Leu | Missense | LP (PM2, PM6, PP2, PP3) | NM_001271.4 | 602119 |
| CHD3 | c.3481C>T | p.His1161Tyr | Missense | LP (PM1, PM2, PP2, PP3) | NM_001005273.3 | 7806365 |
| CHD8 * | c.4987_5003del | p.Val1663Glnfs*59 | Frameshift | P (PVS1, PS2, PM2) | NM_020920.4 | 610528 |
| COL6A3 * | c.6156+2T>C | - | Splicing site | P (PVS1, PS2, PM2, PP3) | NM_004369.4 | 120250 |
| COL9A3 # | c.1021C>T | p.Arg341* | Nonsense | LP (PVS1, PM2) | NM_001853.4 | 120270 |
| CUL3 * | c.769delG | p.Glu257Lysfs*5 | Frameshift | P (PVS1, PS2, PM2) | NM_003590 | 603136 |
| CUL3 * | c.494dup | p.Leu166Ilefs*37 | Frameshift | P (PVS1, PS2, PM2) | NM_003590.4 | 603136 |
| DCX | c.166C>G | p.Arg56Gly | Missense | LP (PM1, PM2, PM5, PP2, PP3) | NM_001195553.1 | 300121 |
| DDX3X | c.1646A>T | p.Asn549Ile | Missense | LP (PM1, PM2, PP2, PP3) | NM_001356.4 | 300160 |
| DNM1 * | c.1751A>T | p.His584Leu | Missense | LP (PS2, PM2) | NM_004408.4 | 602377 |
| DNM1 * | c.2318+2T>C | - | Splicing site | P (PVS1, PS2, PM2) | NM_004408.4 | 602377 |
| DYNC1H1 * | c.11632C>G | p.Gln3878Glu | Missense | LP (PS2, PM2) | NM_001376.5 | 600112 |
| EP300 * | c.4779+1G>A | - | Splicing site | P (PVS1, PS2, PM2) | NM_001429.3 | 602700 |
| FBXO11 * | c.1685A>G | p.Tyr562Cys | Missense | LP (PS2, PM1, PM2, PP2, PP3) | NM_025133.4 | 607871 |
| GNB1 * | c.310G>C | p.Ala104Pro | Missense | LP (PS2, PM2, PP3) | NM_002074.5 | 139380 |
| GRIN1 * | c.2248G>A | p.Gly750Arg | Missense | LP (PS2, PM2, PP2, PP3) | NM_000832.7 | 138249 |
| GRIN1 * | c.1824G>C | p.Trp608Cys | Missense | P (PS2, PM1, PM2, PP2, PP3) | NM_007327.3 | 138249 |
| GRIN2B * | c.1990T>C | p.Ser664Pro | Missense | LP (PS2, PM2, PP3) | NM_000834.3 | 138252 |
| HNRNPU | c.1484_1487del | p.Lys495Ilefs*5 | Frameshift | LP (PVS1, PM2) | NM_031844.3 | 602869 |
| HNRNPU * | c.1576_1580del | p.Asn526Serfs*9 | Frameshift | P (PVS1, PS2, PM2) | NM_031844.2 | 602869 |
| IFIH1 # | c.2863C>T | p.Gln955* | Nonsense | LP (PVS1, PM2) | NM_022168.3 | 606951 |
| JAG1 | c.1725_1726dupTG | p.Asp576Valfs*168 | Frameshift | LP (PVS1, PM2) | NM_000214.3 | 601920 |
| KAT6A * | c.1019del | p.Asn340Thrfs*3 | Frameshift | P (PVS1, PS2, PM2) | NM_006766.4 | 601408 |
| KAT6A * | c.4140_4141insA | p.Asp1381Argfs*13 | Frameshift | P (PVS1, PS2, PM2) | NM_006766.4 | 601408 |
| KCNB1 * | c.1223C>T | p.Pro408Leu | Missense | LP (PS2, PM1, PM2, PP3) | NM_004975.3 | 600397 |
| KCNB1 * | c.1202G>T | p.Gly401Val | Missense | P (PS2, PM1, PM2, PM5, PP3) | NM_004975.4 | 600397 |
| KCNMA1 # | c.2095A>T | p.Lys699* | Nonsense | LP (PVS1, PM2) | NM_001161352.1 | 600150 |
| KCNT2 | c.3118C>T | p.Arg1040* | Nonsense | LP (PVS1, PM2) | NM_198503 | 610044 |
| KDM3B * | c.3638dup | p.(Asp1214Ter | Nonsense | LP (PS2 PM2) | NM_016604.4 | 609373 |
| KDM4B | c.2147del | p.Leu716Tyrfs*42 | Frameshift | LP (PVS1, PM2) | NM_015015.2 | 609765 |
| KDM5C * | c.1571A>T | p.Asn524Ile | Missense | LP (PM1, PM2, PM6, PP3) | NM_004187.5 | 314690 |
| KIAA1109 # | c.4118_4119del | p.Ser1373* | Nonsense | LP (PVS1, PM2) | NM_015312.3 | 611565 |
| KIAA1109 * | c.18T>A | p.Asn6Lys | Missense | LP (PS2, PM2) | NM_015312.3 | 611565 |
| KIAA1109 * | c.4118_4119del | p.Ser1373* | Nonsense | LP (PVS1, PM2) | NM_015312.3 | 611565 |
| KMT2A * | c.7187_7188del | p.Pro2396Argfs*2 | Frameshift | P (PVS1, PS2, PM2) | NM_001197104.2 | 159555 |
| KMT2C * | c.5551C>T | p.Gln1851* | Nonsense | P (PVS1, PS2, PM2) | NM_170606.2 | 606833 |
| KMT2E # | c.1944_1948del | p.Lys649GlufsTer8 | Frameshift | LP (PVS1, PM2) | NM_182931.3 | 608444 |
| LARP7 # | c.1118_1130del | p.Val373Glufs*11 | Frameshift | LP (PVS1, PM2) | NM_016648.4 | 612026 |
| LARS2 * | c.1420del | p.Leu474Trpfs*6 | Frameshift | P (PVS1, PS2, PM2) | NM_015340.4 | 604544 |
| MN1 | c.97del | p.His33ThrfsTer20 | Frameshift | LP (PVS1, PM2) | NM_002430.3 | 156100 |
| NAA15 * | c.2214del | p.Met738IlefsTer18 | Frameshift | P (PVS1, PS2, PM2) | NM_057175.5 | 608000 |
| NACC1 * | c.166C>T | p.Arg56Trp | Missense | LP (PS2, PM2, PP3) | NM_052876.3 | 610672 |
| NEXMIF | c.1998del | p.Glu667Lysfs*5 | Frameshift | LP (PVS1, PM2) | NM_001008537.2 | 300524 |
| NF1 | c.2056A>T | p.Lys686* | Nonsense | LP (PVS1, PM2) | NM_001042492.2 | 613113 |
| NFIB * | c.626A>G | p.Glu209Gly | Missense | LP (PS2, PM2, PP2, PP3) | NM_001190737.3 | 600728 |
| NFIX * | c.442del | p.Ile148Serfs*71 | Frameshift | P (PVS1, PS2, PM2) | NM_001271043.2 | 164005 |
| LPM1D * | c.1245dupT | p.Thr416Tyrfs*18 | Frameshift | P (PVS1, PS2, PM2) | NM_003620.4 | 605100 |
| PUF60 * | c.1334C>T | p.Thr445Ile | Missense | LP (PS2, PM2, PP3) | NM_014281.5 | 604819 |
| RFX7 * | c.2236C>T | p.Gln746* | Nonsense | P (PVS1, PS2, PM2) | NM_022841.7 | 612660 |
| RPS6KA3 * | c.383C>T | p.Pro128Leu | Missense | LP (PM2, PM6, PP2, PP3) | NM_004586.3 | 300075 |
| SATB2 | c.1165C>A | p.Arg389Ser | Missense | LP (PS1, PM2, PM5, PP3) | NM_001172509.2 | 608148 |
| SCN1A * | c.4987G>T | p.Gly1663Cys | Missense | P (PS2, PM1, PM2, PP2, PP3) | NM_006920.6 | 182389 |
| SCN1A * | c.4582-1_4583del | - | Splicing site | P (PVS1, PS2, PM2) | NM_001165963.4 | 182389 |
| SCN1A * | c.4987G>T | p.Gly1663Cys | Missense | P (PS2, PM1, PM2, PP2, PP3) | NM_006920.6 | 182389 |
| SCN2A # | c.641C>A | p.Ser214* | Nonsense | LP (PVS1, PM2) | NM_001040143.2 | 182390 |
| SCN8A * | c.5235C>A | p.Phe1745Leu | Missense | P (PS2, PM1, PM2, PP2, PP3) | NM_001330260 | 600702 |
| SETD1A * | c.5116C>G | p.Leu1706Val | Missense | LP (PS2, PM2) | NM_014712.3 | 611052 |
| SHANK3 * | c.352dup | p.Leu118ProfsTer28 | Frameshift | P (PVS1, PM6, PM2) | NM_001372044.2 | 606230 |
| SIX3 * | c.221del | p.Pro74Argfs*177 | Frameshift | P (PVS1, PM6, PM2) | NM_005413.4 | 603714 |
| SPEN * | c.5485_5486insTTTGAAC | p.Gln1829Leufs*2 | Frameshift | P (PVS1, PS2, PM2) | NM_015001.4 | 613484 |
| STAG2 | c.1018-2_1018-1delinsTT | - | Splicing site | LP (PVS1, PM2) | NM_001042750.2 | 300826 |
| STXBP1 * | c.903-1G>C | - | Splicing site | P (PVS1, PS2, PM2) | NM_001032221.3 | 602926 |
| SYNGAP1 * | c.1713_1714delinsAC | p.Trp572Arg | Missense | LP (PS2, PM2, PM5) | NM_006772.2 | 603384 |
| SYNGAP1 * | c.1216_1218delins | p.Tyr406Asnfs*4 | Frameshift | P (PVS1, PS2, PM2) | NM_006772 | 603384 |
| TAOK1 | c.1721dupA | p.Ser575Glufs*28 | Frameshift | LP (PVS1, PM2) | NM_020791 | 610266 |
| TAOK1 # | c.1489_1492del | p.Asp497Lysfs*42 | Frameshift | LP (PVS1, PM2) | NM_020791.4 | 610266 |
| TBR1 * | c.893dup | p.His298Glnfs*23 | Frameshift | P (PVS1, PS2, PM2) | NM_006593.3 | 604616 |
| TCF20 * | c.5047_5054del | p.Pro1683Valfs*34 | Frameshift | P (PVS1, PS2, PM2) | NM_005650.3 | 603107 |
| TCF4 | c.-20-184_72+815del | - | Splicing site | LP (PVS1, PM2) | NM_001083962.2 | 602272 |
| TNPO3 # | c.120+2T>G | - | Splicing site | LP (PVS1, PM2) | NM_012470.4 | 610032 |
| TRIP12 | c.3206+1G>T | - | Splicing site | LP (PVS1, PM2) | NM_001348323.3 | 604506 |
| TUBB * | c.1145C>T | p.Ser382Leu | Missense | LP (PS2, PM2, PP2, PP3) | NM_178014.4 | 191130 |
| TUBB * | c.1017C>G | p.Ser339Arg | Missense | LP (PS2, PM2, PP2, PP3) | NM_178014.4 | 191130 |
| USP7 * | c.502T>C | p.Ser168Pro | Missense | LP (PS2, PM1, PM2) | NM_003470.2 | 602519 |
| WAC * | c.620del | p.Lys207Serfs*124 | Frameshift | P (PVS1, PS2, PM2) | NM_016628.5 | 615049 |
| ZMIZ1 * | c.1413+4A>G | - | Splicing site | LP (PS2, PM2) | NM_020338.3 | 607159 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamilla, J.; Castro-Cuesta, T.A.; Rueda-Gaitán, P.; Rios Pinto, L.C.; Rodríguez Gutiérrez, D.A.; Sanchez Rubio, Y.N.; Estrada-Serrato, C.; Londoño, O.; Rucinski, C.; Arcos-Burgos, M.; et al. A Robust and Comprehensive Study of the Molecular and Genetic Basis of Neurodevelopmental Delay in a Sample of 3244 Patients, Evaluated by Exome Analysis in a Latin Population. Diagnostics 2025, 15, 376. https://doi.org/10.3390/diagnostics15030376
Lamilla J, Castro-Cuesta TA, Rueda-Gaitán P, Rios Pinto LC, Rodríguez Gutiérrez DA, Sanchez Rubio YN, Estrada-Serrato C, Londoño O, Rucinski C, Arcos-Burgos M, et al. A Robust and Comprehensive Study of the Molecular and Genetic Basis of Neurodevelopmental Delay in a Sample of 3244 Patients, Evaluated by Exome Analysis in a Latin Population. Diagnostics. 2025; 15(3):376. https://doi.org/10.3390/diagnostics15030376
Chicago/Turabian StyleLamilla, Julian, Taryn A. Castro-Cuesta, Paula Rueda-Gaitán, Laura Camila Rios Pinto, Diego Alejandro Rodríguez Gutiérrez, Yuri Natalia Sanchez Rubio, Carlos Estrada-Serrato, Olga Londoño, Cynthia Rucinski, Mauricio Arcos-Burgos, and et al. 2025. "A Robust and Comprehensive Study of the Molecular and Genetic Basis of Neurodevelopmental Delay in a Sample of 3244 Patients, Evaluated by Exome Analysis in a Latin Population" Diagnostics 15, no. 3: 376. https://doi.org/10.3390/diagnostics15030376
APA StyleLamilla, J., Castro-Cuesta, T. A., Rueda-Gaitán, P., Rios Pinto, L. C., Rodríguez Gutiérrez, D. A., Sanchez Rubio, Y. N., Estrada-Serrato, C., Londoño, O., Rucinski, C., Arcos-Burgos, M., Isaza-Ruget, M., & López Rivera, J. J. (2025). A Robust and Comprehensive Study of the Molecular and Genetic Basis of Neurodevelopmental Delay in a Sample of 3244 Patients, Evaluated by Exome Analysis in a Latin Population. Diagnostics, 15(3), 376. https://doi.org/10.3390/diagnostics15030376