
Ganciclovir Resistance-Linked Mutations in the HCMV UL97 Gene: Sanger Sequencing Analysis in Samples from Transplant Recipients at a Tertiary Hospital in Southern Brazil

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples and Ethical Considerations
2.2. DNA Extraction and Polymerase Chain Reaction (PCR) Amplification
2.3. In Silico Analysis
2.4. Limit of Detection (LoD) and Repeatability
2.5. Bi-Directional Sanger Sequencing
2.6. Data and Mutation Analysis
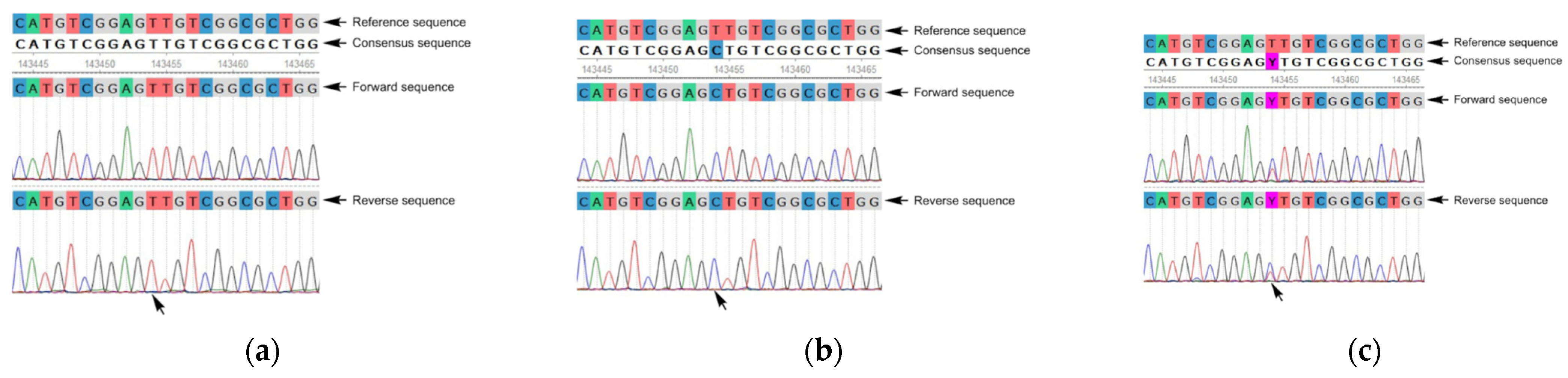
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-Organ Transplantation. Transplantation 2018, 102, 900–931. [Google Scholar] [CrossRef] [PubMed]
- Kotton, C.N.; Kamar, N. New Insights on CMV Management in Solid Organ Transplant Patients: Prevention, Treatment, and Management of Resistant/Refractory Disease. Infect Dis. Ther. 2023, 12, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Gorenec, L. Human Cytomegalovirus (HCMV) Genetic Diversity, Drug Resistance Testing and Prevalence of the Resistance Mutations: A Literature Review. TropicalMed 2024, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.A.; Passos, A.M.; Treitinger, A.; Spada, C. Seroprevalence of Cytomegalovirus Antibodies in Blood Donors in Southern, Brazil. Rev. Soc. Bras. Med. Trop. 2010, 43, 359–361. [Google Scholar] [CrossRef]
- Oliveira, A.; Nunes, G.; Sousajr, I.; Gonçalves, J.; Lopes, J.; Silva, C.; Silva, J.; Amorim, L.; Paula, V. Prevalência Molecular e Sorológica do Citomegalovírus Humano em Doadores de Sangue do Rio de Janeiro. Hematol. Transfus. Cell Ther. 2022, 44, S648. [Google Scholar] [CrossRef]
- Avery, R.K.; Alain, S.; Alexander, B.D.; Blumberg, E.A.; Chemaly, R.F.; Cordonnier, C.; Duarte, R.F.; Florescu, D.F.; Kamar, N.; Kumar, D.; et al. Maribavir for Refractory Cytomegalovirus Infections With or Without Resistance Post-Transplant: Results From a Phase 3 Randomized Clinical Trial. Clin. Infect. Dis. 2022, 75, 690–701. [Google Scholar] [CrossRef]
- Chou, S.; Alain, S.; Cervera, C.; Chemaly, R.F.; Kotton, C.N.; Lundgren, J.; Papanicolaou, G.A.; Pereira, M.R.; Wu, J.J.; Murray, R.A.; et al. Drug Resistance Assessed in a Phase 3 Clinical Trial of Maribavir Therapy for Refractory or Resistant Cytomegalovirus Infection in Transplant Recipients. J. Infect. Dis. 2024, 229, 413–421. [Google Scholar] [CrossRef]
- Tasoujlu, M.; Khalafkhany, D.; Makhdoomi, K.; Motazakker, M. Cytomegalovirus UL97 Ganciclovir Resistance Mutations in Kidney Transplant Recipients. Bratisl. Lek. Listy 2022, 123, 518–522. [Google Scholar] [CrossRef]
- Silva, D.D.F.L.D.; Cardoso, J.F.; Silva, S.P.D.; Arruda, L.M.F.; Medeiros, R.L.F.D.; Moraes, M.M.; Sousa, R.C.M. HCMV UL97 Phosphotransferase Gene Mutations May Be Associated with Antiviral Resistance in Immunocompromised Patients in Belém, PA, Northern Brazil. Rev. Soc. Bras. Med. Trop. 2018, 51, 141–145. [Google Scholar] [CrossRef]
- Lurain, N.S.; Chou, S. Antiviral Drug Resistance of Human Cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef]
- Chou, S.; Bowlin, T.L. Cytomegalovirus UL97 Mutations Affecting Cyclopropavir and Ganciclovir Susceptibility. Antimicrob Agents Chemother 2011, 55, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R. Drug-Resistant Cytomegalovirus: Clinical Implications of Specific Mutations. Curr. Opin. Organ Transplant. 2018, 23, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Watters, M.; Sinha, R.; Kleiboeker, S. Ganciclovir and Maribavir Susceptibility Phenotypes of Cytomegalovirus UL97 ATP Binding Region Mutations Detected by Expanded Genotypic Testing. Antivir. Res. 2021, 193, 105139. [Google Scholar] [CrossRef] [PubMed]
- Alsanea, M.S.; Al-Qahtani, A.A.; Almaghrabi, R.S.; AlAbdulkareem, M.A.; Alahideb, B.M.; Obeid, D.; Alsuwairi, F.A.; Alhamlan, F.S. Diagnosis of Human Cytomegalovirus Drug Resistance Mutations in Solid Organ Transplant Recipients—A Review. Diagnostics 2024, 14, 203. [Google Scholar] [CrossRef]
- Grossi, P.A.; Peghin, M. Recent Advances in Cytomegalovirus Infection Management in Solid Organ Transplant Recipients. Curr. Opin. Organ Transplant. 2024, 29, 131–137. [Google Scholar] [CrossRef]
- Babloyan, S.; Voskanyan, M.; Shekherdimian, S.; Nazaryan, H.; Arakelyan, S.; Kurkchyan, K.; Geyikyan, P.; Babloyan, A.; Sarkissian, A. Kidney Transplantation in Low- to Middle-Income Countries: Outcomes and Lessons Learned from Armenia. Ann Transpl. 2021, 26, e930943-1. [Google Scholar] [CrossRef]
- Panda, K.; Parashar, D.; Viswanathan, R. An Update on Current Antiviral Strategies to Combat Human Cytomegalovirus Infection. Viruses 2023, 15, 1358. [Google Scholar] [CrossRef]
- Fryer, J.F.; Heath, A.B.; Minor, P.D.; Kessler, H.; Rawlinson, W.; Boivin, G.; Preiksaitis, J.; Pang, X.-L.; Barranger, C.; Alain, S.; et al. A Collaborative Study to Establish the 1st WHO International Standard for Human Cytomegalovirus for Nucleic Acid Amplification Technology. Biologicals 2016, 44, 242–251. [Google Scholar] [CrossRef]
- Jerome, K.R.; Huang, M.-L.; Wald, A.; Selke, S.; Corey, L. Quantitative Stability of DNA after Extended Storage of Clinical Specimens as Determined by Real-Time PCR. J. Clin. Microbiol. 2002, 40, 2609–2611. [Google Scholar] [CrossRef]
- Martí-Carreras, J.; Maes, P. Human Cytomegalovirus Genomics and Transcriptomics through the Lens of Next-Generation Sequencing: Revision and Future Challenges. Virus Genes 2019, 55, 138–164. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Crossley, B.M.; Bai, J.; Glaser, A.; Maes, R.; Porter, E.; Killian, M.L.; Clement, T.; Toohey-Kurth, K. Guidelines for Sanger Sequencing and Molecular Assay Monitoring. J. VET Diagn. Investig. 2020, 32, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Tilloy, V.; Díaz-González, D.; Laplace, L.; Bisserier, E.; Chou, S.; Rawlinson, W.D.; Boivin, G.; Baldanti, F.; Lazzarotto, T.; Andrei, G.; et al. Comprehensive Herpesviruses Antiviral Drug Resistance Mutation Database (CHARMD). Antivir. Res. 2024, 231, 106016. [Google Scholar] [CrossRef] [PubMed]
- Chevillotte, M.; Von Einem, J.; Meier, B.M.; Lin, F.-M.; Kestler, H.A.; Mertens, T. A New Tool Linking Human Cytomegalovirus Drug Resistance Mutations to Resistance Phenotypes. Antivir. Res. 2010, 85, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Castor, J.; Cook, L.; Corey, L.; Jerome, K.R. Rapid Detection Directly from Patient Serum Samples of Human Cytomegalovirus UL97 Mutations Conferring Ganciclovir Resistance. J. Clin. Microbiol. 2007, 45, 2681–2683. [Google Scholar] [CrossRef]
- Tamura, S.; Osawa, S.; Ishida, N.; Miyazu, T.; Tani, S.; Yamade, M.; Iwaizumi, M.; Hamaya, Y.; Kosugi, I.; Furuta, T.; et al. Prevalence of UL97 Gene Mutations and Polymorphisms in Cytomegalovirus Infection in the Colon Associated with or without Ulcerative Colitis. Sci. Rep. 2021, 11, 13676. [Google Scholar] [CrossRef]
- Chorlton, S.D.; Ritchie, G.; Lawson, T.; McLachlan, E.; Romney, M.G.; Matic, N.; Lowe, C.F. Next-Generation Sequencing for Cytomegalovirus Antiviral Resistance Genotyping in a Clinical Virology Laboratory. Antivir. Res. 2021, 192, 105123. [Google Scholar] [CrossRef]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human Cytomegalovirus Clinical Isolates Carry at Least 19 Genes Not Found in Laboratory Strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [CrossRef]
- Vo, M.; Aguiar, A.; McVoy, M.A.; Hertel, L. Cytomegalovirus Strain TB40/E Restrictions and Adaptations to Growth in ARPE-19 Epithelial Cells. Microorganisms 2020, 8, 615. [Google Scholar] [CrossRef]
- Burd, E.M. Validation of Laboratory-Developed Molecular Assays for Infectious Diseases. Clin. Microbiol. Rev. 2010, 23, 550–576. [Google Scholar] [CrossRef]
- Mallory, M.A.; Hymas, W.C.; Simmon, K.E.; Pyne, M.T.; Stevenson, J.B.; Barker, A.P.; Hillyard, D.R.; Hanson, K.E. Development and Validation of a Next-Generation Sequencing Assay with Open-Access Analysis Software for Detecting Resistance-Associated Mutations in CMV. J. Clin. Microbiol. 2023, 61, e00829-23. [Google Scholar] [CrossRef] [PubMed]
- Streck, N.T.; Espy, M.J.; Ferber, M.J.; Klee, E.W.; Razonable, R.R.; Gonzalez, D.; Sayada, C.; Heaton, P.R.; Chou, S.; Binnicker, M.J. Use of Next-Generation Sequencing to Detect Mutations Associated with Antiviral Drug Resistance in Cytomegalovirus. J. Clin. Microbiol. 2023, 61, e00429-23. [Google Scholar] [CrossRef] [PubMed]
- Chou, S. Recombinant Phenotyping of Cytomegalovirus UL97 Kinase Sequence Variants for Ganciclovir Resistance. Antimicrob Agents Chemother 2010, 54, 2371–2378. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Ercolani, R.J.; Vanarsdall, A.L. Differentiated Levels of Ganciclovir Resistance Conferred by Mutations at Codons 591 to 603 of the Cytomegalovirus UL97 Kinase Gene. J Clin Microbiol 2017, 55, 2098–2104. [Google Scholar] [CrossRef]
- Recio, V.; González, I.; Tarragó, D. Cytomegalovirus Drug Resistance Mutations in Transplant Recipients with Suspected Resistance. Virol. J. 2023, 20, 153. [Google Scholar] [CrossRef]
- Chou, S.; Watanabe, J. Ganciclovir and Maribavir Cross-Resistance Revisited: Relative Drug Susceptibilities of Canonical Cytomegalovirus Mutants. Antivir. Res. 2024, 222, 105792. [Google Scholar] [CrossRef]
- Steingruber, M.; Marschall, M. The Cytomegalovirus Protein Kinase pUL97: Host Interactions, Regulatory Mechanisms and Antiviral Drug Targeting. Microorganisms 2020, 8, 515. [Google Scholar] [CrossRef]
- Erice, A.; Borrell, N.; Li, W.; Miller, W.J.; Balfour, H.H. Ganciclovir Susceptibilities and Analysis of UL97 Region in Cytomegalovirus (CMV) Isolates from Bone Marrow Recipients with CMV Disease after Antiviral Prophylaxis. J. Infect. Dis. 1998, 178, 531–534. [Google Scholar] [CrossRef]
- Tanaka, K.; Hori, T.; Yoto, Y.; Hatakeyama, N.; Yamamoto, M.; Suzuki, N.; Tsutsumi, H. Human Cytomegalovirus UL97 D605E Polymorphism Has a High Prevalence in Immunocompetent Japanese Infants and Children. Microbiol. Immunol. 2011, 55, 328–330. [Google Scholar] [CrossRef]
- Arav-Boger, R. Strain Variation and Disease Severity in Congenital Cytomegalovirus Infection. Infect. Dis. Clin. North Am. 2015, 29, 401–414. [Google Scholar] [CrossRef]
- Puchhammer-Stöckl, E.; Görzer, I.; Zoufaly, A.; Jaksch, P.; Bauer, C.C.; Klepetko, W.; Popow-Kraupp, T. Emergence of Multiple Cytomegalovirus Strains in Blood and Lung of Lung Transplant Recipients. Transplantation 2006, 81, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Görzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stöckl, E. Deep Sequencing Reveals Highly Complex Dynamics of Human Cytomegalovirus Genotypes in Transplant Patients over Time. J. Virol. 2010, 84, 7195–7203. [Google Scholar] [CrossRef] [PubMed]
- Royston, L.; Papanicolaou, G.A.; Neofytos, D. Refractory/Resistant Cytomegalovirus Infection in Transplant Recipients: An Update. Viruses 2024, 16, 1085. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Sample ID | Transplant Type | Viral Load (Copies/mL; IU/mL) | Period of Collection | Method of DNA Extraction |
|---|---|---|---|---|
| A2 | Kidney | 3272; 5104 | 07/2022 | m2000 |
| A4 | Kidney | 8304; 12,954 | 07/2022 | m2000 |
| A5 | Kidney | 2964; 4624 | 07/2022 | m2000 |
| A6 | Kidney | 4994; 7791 | 07/2022 | m2000 |
| A10 | Liver | 46,544; 76,798 | 08/2022 | m2000 |
| A17 | Liver | 3424; 5341 | 09/2022 | m2000 |
| A20 | Bone marrow | 2962; 4621 | 09/2022 | m2000 |
| A21 | Heart and kidney | 108,903; 169,889 | 09/2022 | m2000 |
| A26 | Bone marrow | 6196; 9666 | 09/2022 | m2000 |
| A37 | Kidney | 6795; 10,600 | 11/2022 | QIAmp |
| A39 | Liver | 13,510; 21,076 | 11/2022 | QIAmp |
| A44 | Kidney | 156,554; 244,224 | 11/2022 | QIAmp |
| A46 | Kidney | 121,222; 189,106 | 01/2023 | QIAmp |
| A48 | Bone marrow | 86,809; 135,422 | 03/2023 | QIAmp |
| A53 | Bone marrow | 65,958; 102,894 | 04/2024 | QIAmp |
| A54 | Bone marrow | 111,697; 174,247 | 06/2024 | QIAmp |
| PCR Round | Primer | Sequence (5′-3′) | Size (bp) |
|---|---|---|---|
| First reaction | 1278F | GGCGTGCATCGACAGCTACCG | 736 |
| 2013R | CCGTCGCTCGTGGCCAACAGA | ||
| Second reaction | 1292F | GCTACCGACGTGCCTTTTGCA | 707 |
| 1998R | AACAGACGCTCCACGTTCTTT |
| Sample ID | Genomic Annotation | Amino Acid Change | GCV Resistance |
|---|---|---|---|
| A4 | A143.326G | N510S | Genetic polymorphism 1 |
| A5 | C143.323T | P509L | Unknown mutation 2 |
| A6 | C143.578T | A594V | Mutation associated with drug resistance 3 |
| C143.781T | H662Y | Unknown mutation 2 | |
| A17 | C143.612G | D605E | Genetic polymorphism 1 |
| A44 | C143.578T | A594V | Mutation associated with drug resistance 3 |
| C143.606G | C603W | Mutation associated with drug resistance 3 | |
| A48 | C143.606G | C603W | Mutation associated with drug resistance 3 |
| A53 | C143.578T | A594V | Mutation associated with drug resistance 3 |
| A54 | G143.679A | A628T | Unknown mutation 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Rocha, A.C.A.; Rodrigues, G.M.; da Silva Hellwig, A.H.; Pereira, D.C.; Volpato, F.C.Z.; Barth, A.L.; de-Paris, F. Ganciclovir Resistance-Linked Mutations in the HCMV UL97 Gene: Sanger Sequencing Analysis in Samples from Transplant Recipients at a Tertiary Hospital in Southern Brazil. Diagnostics 2025, 15, 214. https://doi.org/10.3390/diagnostics15020214
da Rocha ACA, Rodrigues GM, da Silva Hellwig AH, Pereira DC, Volpato FCZ, Barth AL, de-Paris F. Ganciclovir Resistance-Linked Mutations in the HCMV UL97 Gene: Sanger Sequencing Analysis in Samples from Transplant Recipients at a Tertiary Hospital in Southern Brazil. Diagnostics. 2025; 15(2):214. https://doi.org/10.3390/diagnostics15020214
Chicago/Turabian Styleda Rocha, Anna Caroline Avila, Grazielle Motta Rodrigues, Alessandra Helena da Silva Hellwig, Dariane Castro Pereira, Fabiana Caroline Zempulski Volpato, Afonso Luís Barth, and Fernanda de-Paris. 2025. "Ganciclovir Resistance-Linked Mutations in the HCMV UL97 Gene: Sanger Sequencing Analysis in Samples from Transplant Recipients at a Tertiary Hospital in Southern Brazil" Diagnostics 15, no. 2: 214. https://doi.org/10.3390/diagnostics15020214
APA Styleda Rocha, A. C. A., Rodrigues, G. M., da Silva Hellwig, A. H., Pereira, D. C., Volpato, F. C. Z., Barth, A. L., & de-Paris, F. (2025). Ganciclovir Resistance-Linked Mutations in the HCMV UL97 Gene: Sanger Sequencing Analysis in Samples from Transplant Recipients at a Tertiary Hospital in Southern Brazil. Diagnostics, 15(2), 214. https://doi.org/10.3390/diagnostics15020214