1. Introduction
Wolfram syndrome (WS), also known as DIDMOAD (Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, and Deafness), is a rare autosomal recessive neurodegenerative disorder affecting multiple systems. Its prevalence is estimated at 1 in 770,000 individuals in the United Kingdom and approximately 1 in 1 million in Spain [
1,
2]. WS is characterized by early-onset, non-autoimmune insulin-dependent diabetes mellitus and optic atrophy, with subsequent development of diabetes insipidus and sensorineural hearing loss [
3,
4,
5,
6,
7]. Gonadal dysfunction, though less studied, may also significantly affect patients’ quality of life and contribute to the broader phenotype of the disease [
8].
Recent studies suggest that gonadal dysfunction could serve as a relevant diagnostic criterion for WS, particularly in pediatric and young adult patients, underscoring the need for systematic screening and early intervention [
9,
10,
11]. Hypogonadism in WS may manifest as either hypergonadotropic or hypogonadotropic, affecting both males and females [
12,
13]. In males, primary testicular dysfunction is the predominant form, associated with low testosterone levels and histological findings such as seminiferous tubule hyalinization and Leydig cell depletion [
12,
13,
14]. Genetic underpinnings of hypogonadism, including congenital hypogonadotropic forms, are increasingly recognized in syndromic presentations like WS [
14,
15,
16].
Hypogonadism in males with WS may be classified as primary (low testosterone with high LH/FSH) or secondary (low testosterone with low or normal LH/FSH) [
12,
13,
14,
15,
16]. Diagnostic criteria include total testosterone levels below 300 ng/dL (10.4 nmol/L), preferably measured in the morning [
15]. When SHBG alterations are suspected, free testosterone should also be evaluated [
16]. Clinical symptoms include reduced libido, erectile dysfunction, fatigue, muscle loss, and osteoporosis [
16,
17]. The differential diagnosis is further supported by LH and FSH levels, although not diagnostic on their own [
15]. Recent guidelines emphasize evaluating both functional and organic causes of hypogonadism in rare syndromes [
18].
In females, gonadal dysfunction may also be primary or secondary but is harder to detect due to variable hormonal patterns and masking effects of contraceptive use [
11,
19,
20,
21,
22]. Consequently, ovarian insufficiency in WS may go undetected without specific testing [
21,
22]. AMH measurement and imaging are emerging tools for assessing ovarian reserve [
19,
22,
23]. Some studies advocate for including gonadal dysfunction as a diagnostic feature of WS, alongside urinary abnormalities [
10,
23,
24].
Reproductive dysfunction in WS can affect metabolic health, bone density, and psychological well-being [
7,
8,
24]. Symptoms such as fatigue, weakness, cognitive impairment, and mood disorders are often related to hypogonadism [
7,
24]. These findings support early hormonal assessment and timely intervention [
7]. Although treatments such as testosterone replacement are described in guidelines [
15,
18,
25], they are often underused in WS. Recent cohort studies confirm the high prevalence and variability of gonadal dysfunction in WFS1-spectrum disorders, highlighting the need for structured evaluation [
26,
27,
28].
This study is prospective, aiming to evaluate gonadal function in a cohort of patients with genetically confirmed WS, with special attention to clinical, biochemical, and sex-specific features, to promote early recognition and improve clinical management.
2. Materials and Methods
Study Design and Participants: This was a prospective cohort study conducted between 1999 and 2024. A total of 45 patients with genetically confirmed Wolfram syndrome (WS) were enrolled in Spain and Portugal, comprising 25 males and 20 females. At the time of data analysis, 10 patients had died, leaving 35 survivors with a mean age of 26.23 years (SD 9.61). All participants carried pathogenic variants in the WFS1 gene, confirmed by Sanger sequencing (BigDye™ Terminator v3.1 Cycle Sequencing Kit; Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) or, in later cases, next-generation sequencing (NGS) multigene panels run on a MiSeq® platform (Illumina Inc., San Diego, CA, USA) for rare endocrine syndromes.
Inclusion and Exclusion Criteria: Inclusion criteria were: (1) clinical diagnosis of WS, and (2) genetic confirmation of pathogenic or likely pathogenic variants in WFS1, classified according to ACMG/AMP guidelines, detected by Sanger sequencing or multigene next-generation sequencing (NGS). Patients who did not meet both criteria were excluded.
Clinical and Laboratory Evaluation: All patients were systematically evaluated for gonadal dysfunction. Since diabetes mellitus and optic atrophy are core diagnostic features of WS, their presence was documented in every patient. Diabetes diagnosis included screening for autoantibodies (ICA, IAA, GADA, IA-2A; Euroimmun AG, Lübeck, Germany) to confirm the non-autoimmune nature of WS-related diabetes. ZNT8 antibodies were not measured, as they were not included in the standard screening protocols during the earlier years of the study.
Male Evaluation: In males, hormonal profiling included total testosterone, free testosterone, luteinizing hormone (LH), follicle-stimulating hormone (FSH), inhibin B, sex hormone-binding globulin (SHBG), and albumin. Hormone levels were measured using Immulite
® 2000 (Diagnostic Products Corporation, Los Angeles, CA, USA; 1999–2006), ADVIA Centaur
® XP (Siemens Healthineers, Erlangen, Germany; 2007–2016) and Cobas
® e 801 (Roche Diagnostics GmbH, Mannheim, Germany; 2017–2024), with appropriate calibrations across time periods. Semen analysis followed WHO guidelines and assessed sperm concentration, motility, and morphology. Imaging with testicular ultrasound and/or MRI evaluated structural abnormalities. In selected patients, testicular biopsy was performed for histological assessment of seminiferous tubules, Sertoli, and Leydig cell populations. Diagnostic classification of hypogonadism in male patients was based on testosterone and gonadotropin levels, as summarized in
Table 1.
Female Evaluation: In females, data collected included age at menarche, menstrual regularity, dysmenorrhea, and age at menopause. Hormonal evaluation included anti-Müllerian hormone (AMH), FSH, LH, and estradiol, performed using Beckman Coulter or Roche platforms, with assay-specific cutoffs. Pelvic ultrasound assessed ovarian volume, atrophy, and uterine morphology.
Classification of Hypogonadism: Hypogonadism was defined and classified as either primary (hypergonadotropic) or secondary (hypogonadotropic) based on LH and FSH levels. In males, testicular volume was assessed by orchidometry or ultrasounds. Hormonal cutoffs followed international guidelines for age and sex.
Data Collection: Patients were recruited across Spain and Portugal. Evaluations were performed either during scheduled multidisciplinary visits or home visits conducted by the principal investigator.
Study Design, Compliance with Ethical Standards, and Data Analysis
This study was a quantitative, observational, descriptive, and cross-sectional analysis conducted in genetically confirmed WS patients from Spain, with the inclusion of two additional patients from Portugal. The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Torrecárdenas University Hospital (code 75/2020), approval date 27 February 2020.
Prior to study inclusion, written informed consent was obtained from all participants or their legal guardians. For participants under 18 years of age or those unable to provide informed consent, consent was obtained from a parent or legal guardian. Confidentiality and data protection were ensured in accordance with Spanish Organic Law 3/2018, of 5 December, on the Protection of Personal Data and Guarantee of Digital Rights (Spain), BOE No. 294, 6 December 2018, 119788–119857.
Data analysis: Statistical analyses were performed using Python 3.0 statistical libraries, ensuring robust and reproducible computations. Descriptive statistics were applied to all variables, with continuous variables summarized as means ± standard deviations (SD) for normally distributed data and medians with interquartile ranges for non-normally distributed data. Categorical variables were expressed as frequencies and percentages. Correlation analyses were conducted to assess the relationships between hormonal and metabolic biomarkers, age of onset, and severity, identifying statistically significant differences between groups (p < 0.05) and relevant associations.
3. Results
Diabetes mellitus was present in all cases except one patient with an autosomal dominant WFS1-related disorder. In those with diabetes, the onset occurred before the age of 16 years. In 100% of cases, the diabetes was non-autoimmune, with negative autoantibodies (ICA, IAA, GADA, IA-2A). Optic atrophy was also diagnosed before age 16 in all patients, as confirmed by an ophthalmologist. The patient with the dominant form of WFS1, aged 13 years, did not show clinical or biochemical signs of hypogonadism at the time of evaluation.
3.1. Male Patients
3.1.1. General Statistics
Table 2 shows the statistics of the whole population of male patients. Among the 25 male patients, the average luteinizing hormone (LH) level was 12.40 IU/L (SD, 6.91), the average follicle-stimulating hormone (FSH) level was 18.44 IU/L (SD, 13.50), the average albumin level was 4.21 g/dL (SD, 0.52) and the average Sex Hormone-Binding Globulin (SHBG) level was 59.88 g/dL (SD, 26.95). Mean total testosterone was 9.88 nmol/L (SD, 7.06), and free testosterone averaged 0.135 nmol/L (SD, 0.079).
Figure 1 shows the correlation matrix between these biomarkers for the whole male population. A strong positive correlation can be observed between total testosterone and free testosterone(0.83) and a positive correlation between FSH and LH (0.73). The rest of the biomarkers are weakly correlated (positive or negative) or are uncorrelated.
3.1.2. Males with Hypogonadism
In total, 19 male patients met the criteria for hypogonadism. The mean age of onset was 18.35 years (range, 15–29), with the majority developing symptoms during adolescence. Most cases were classified as severe. The mean total testosterone level was 7.29 nmol/L (range, 2.76–19.06), and the mean free testosterone level was 0.103 nmol/L (range, 0.026–0.239), both indicating consistently low androgen levels. LH values ranged from 1.2 to 25.0 IU/L, with a mean of 13.99 IU/L, reflecting variable hypothalamic–pituitary–gonadal axis activity. FSH values ranged from 0.70 to 42.65 IU/L, with a mean of 21.70 IU/L. Mean blood albumin was 4.07 g/dL, within normal range. SHBG levels ranged widely (5.69–147.00 nmol/L), suggesting differences in testosterone bioavailability. The mean SHBG was 59.23 nmol/L.
Table 3 presents the summary statistics for this subgroup.
The correlation matrix between these biomarkers for the population with hypogonadism is presented in
Figure 2. We also analyzed the correlation among these biomarkers, the onset age, and the severity provided by medical examination.
Semen analysis revealed azoospermia in 12 patients (
Table 4). Among the remaining five, two had oligozoospermia, one had isolated teratozoospermia, one had asthenozoospermia, and one showed combined oligoasthenoteratozoospermia. Testicular biopsies were performed in five patients, revealing seminiferous tubule hyalinization, Sertoli cell predominance, and reduced Leydig cells.
3.1.3. Population Without Hypogonadism
Table 5 shows the main biomarkers statistics in this group. This group demonstrated consistently higher androgen levels compared to the hypogonadal subgroup. The mean total testosterone level was 18.08 nmol/L, notably higher than the 6.36 nmol/L observed in the hypogonadism group. Free testosterone averaged 0.234 nmol/L, also significantly elevated relative to the hypogonadal mean of 0.091 nmol/L. Luteinizing hormone (LH) levels averaged 7.35 IU/L (range, 2.8–14.2), which was lower than the mean of 14.37 IU/L in the hypogonadism group, indicating distinct hypothalamic–pituitary–gonadal axis activity. FSH values ranged from 2.39 to 20.30 IU/L, with a mean of 8.10 IU/L. Mean serum albumin was 4.63 g/dL, slightly higher than the 4.07 g/dL found in the hypogonadal group. The mean SHBG level was 61.93 nmol/L, only slightly greater than the 59.23 nmol/L observed in the hypogonadism group.
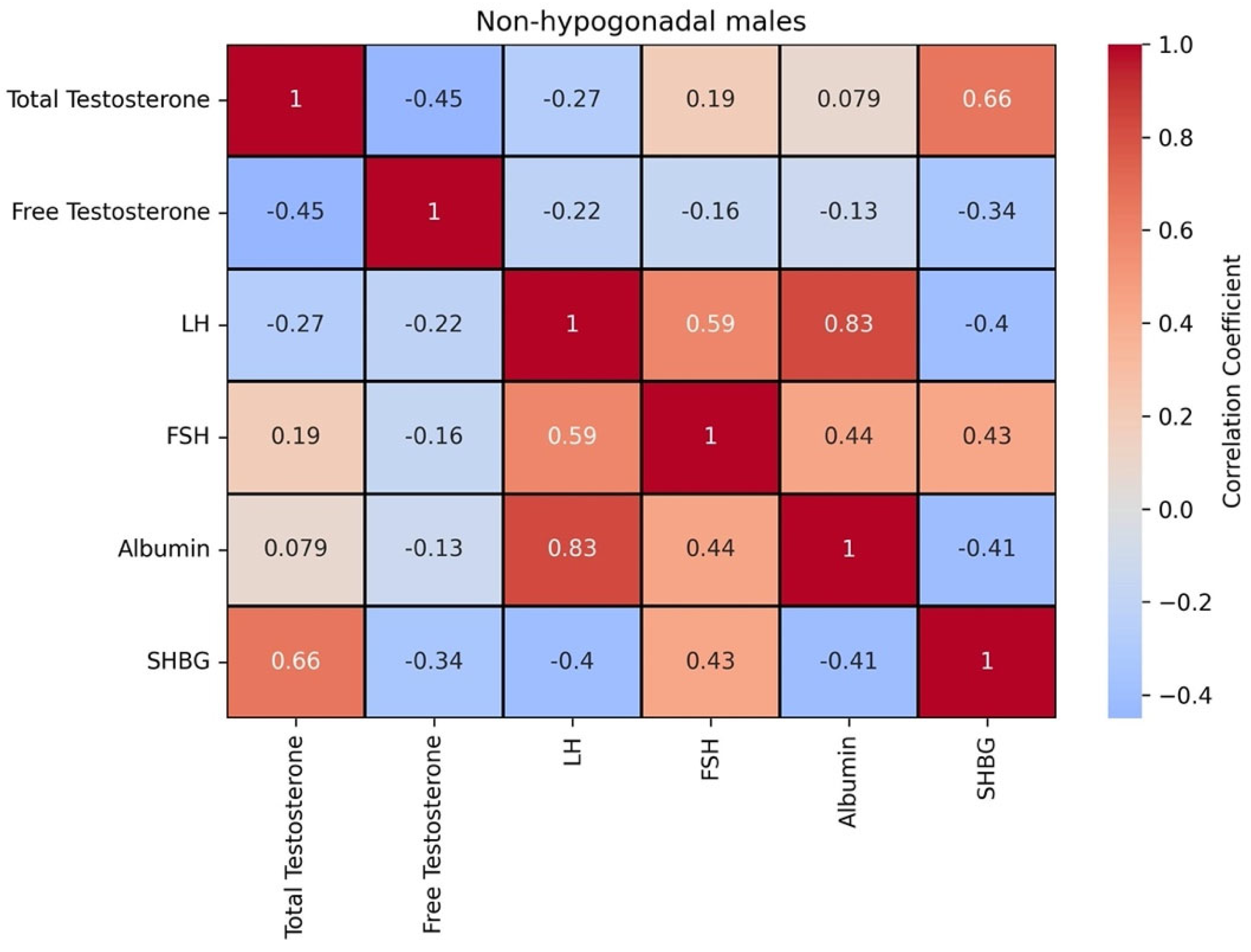
Figure 3 shows the correlation matrix between these biomarkers for the population without hypogonadism.
3.1.4. Analysis of Statistical Differences Between Distributions
Table 6 shows the comparison of hormonal and biochemical markers between patients with and without hypogonadism and the result of the T-test performed on these biomarkers, ordered by statistical significance. The T-value measures how different the group means are relative to the variation in the data. A larger absolute t-value suggests a bigger difference between the groups. The
p-value measures the statistical difference between the biomarkers in both groups; that is, is the difference between the two distributions of these biomarkers likely due to random chance, or is it statistically meaningful? Small
p-values imply that the Null hypothesis (the samples come from the same distribution) should be rejected.
3.1.5. Comparison of Hormonal Profiles
As can be observed in
Table 6, all the biomarkers except SHBG show a significant difference between both groups, with free testosterone and total testosterone being the most significant. Differences in albumin, FSH, and LH are also statistically different at a level of 0.05. The group without hypogonadism exhibited significantly higher levels of total and free testosterone compared to those with hypogonadism. Luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels were markedly elevated in the hypogonadism group, consistent with hypergonadotropic hypogonadism and impaired negative feedback regulation of the hypothalamic–pituitary–gonadal axis. These findings contrast with the eugonadal group, where lower LH and FSH values reflect preserved endocrine feedback. While albumin and SHBG levels were slightly higher in the eugonadal group, these differences were minor and unlikely to meaningfully influence testosterone bioavailability. Notably, testicular volume was less than 12 mL in all the patients with hypogonadism, supporting the diagnosis of primary testicular failure.
3.1.6. Biomarker Correlations
In the non-hypogonadism group, total testosterone and SHBG exhibited a strong positive correlation, while free testosterone and SHBG demonstrated a moderate negative correlation. This may reflect distinct regulatory mechanisms for bound versus free forms of testosterone. LH and albumin were also strongly positively correlated, suggesting potential shared regulatory influences. FSH is positively correlated with all the biomarkers, except for free testosterone. These findings, along with the moderate inverse relationship between SHBG and free testosterone, underscore the relevance of SHBG in modulating testosterone bioavailability (
Figure 3). The correlation between these biomarkers with the onset age and the severity shows that the former is positively correlated to SHBG (0.68) and the latter is negatively correlated to total testosterone (−0.73) and free testosterone (−0.74).
In contrast, within the hypogonadism group, the strongest correlations were observed between total testosterone and free testosterone (0.9), between LH and FSH (0.68), and total testosterone and SHBG (0.44). SHBG displayed negative weak correlations with FSH (−0.55) and LH (−0.46). Albumin showed weak positive correlations with both total and free testosterone but was not strongly associated with other variables (
Figure 2).
These findings might obviously be affected by data scarcity and should be interpreted with caution. Nevertheless, these correlations seem to be very logical.
3.2. Female Patients
All 20 female patients experienced menstrual irregularities requiring hormonal therapy. The mean age at menarche was 14 years (SD, 1.57). Four patients underwent menopause at a mean age of 35 years (SD, 5.56), with the earliest case occurring at 27 years. Only one woman had given birth at the age of 25. Menstrual disturbances were common, and 20% of the cohort experienced premature menopause. Four women had low anti-Müllerian hormone (AMH) levels (≤0.53 ng/mL). The mean AMH level was 0.30 ng/mL (SD, 0.21), and the median was 0.35 ng/mL. The average age of participants was 23 years (SD, 4.32), with a median age of 24 years.
Notably, the patient with the highest AMH level (0.5 ng/mL) was 17 years old, while the lowest level (0.01 ng/mL) was recorded in a 29-year-old woman. Ultrasound showed ovarian atrophy in three cases and a menopausal-appearing uterus in two of them. Remarkably, the woman with the lowest AMH level achieved embryo implantation via oocyte donation after 14 months of hormonal therapy to reach appropriate endometrial thickness. These findings indicate a predisposition to hypogonadism or earlier ovarian dysfunction compared to the general population (
Table 7).
4. Discussion
Our study reveals a strikingly high prevalence of gonadal dysfunction in patients with Wolfram syndrome (WS), particularly among males. Hypogonadism was detected in 76.0% of male participants, with 89.5% of these cases classified as hypergonadotropic, characterized by low testosterone levels and elevated LH and FSH. These hormonal profiles, together with consistent histological findings—such as seminiferous tubule hyalinization, Sertoli cell predominance, and Leydig cell depletion—strongly suggest that primary testicular failure is the dominant pathophysiological mechanism, in agreement with previous studies [
5,
9,
12,
13].
Reproductive function was severely impaired: 70.6% of hypogonadal males had azoospermia, while the rest exhibited oligozoospermia, teratozoospermia, or asthenozoospermia. This degree of spermatogenic failure reinforces the importance of early fertility assessment and individualized reproductive counseling in WS males. Interestingly, in the subgroup without hypogonadism, total testosterone averaged 18.08 nmol/L versus 6.36 nmol/L in hypogonadal patients, while LH levels were significantly lower (7.35 vs. 14.37 IU/L), supporting an intact hypothalamic–pituitary–gonadal axis in the former group.
In contrast, the female cohort presents a more nuanced picture. All 20 women experienced menstrual irregularities, and 20% had premature menopause. Although hormonal contraceptive use may obscure clinical features of ovarian insufficiency, our findings—such as AMH levels ≤ 0.53 ng/mL in four patients, ovarian atrophy in three, and a postmenopausal uterus in two—indicate a reduced ovarian reserve. Notably, one patient with an AMH of 0.01 ng/mL and ultrasound evidence of atrophy achieved a pregnancy with donor oocytes after 14 months of hormonal therapy, highlighting the complex reproductive challenges in this group [
19,
20].
Our correlation analyses further support the existence of divergent endocrine profiles between hypogonadal and non-hypogonadal patients. In non-hypogonadal males, testosterone showed a strong positive correlation with SHBG, while free testosterone had a moderate inverse correlation with SHBG—pointing to different regulatory dynamics of free and bound androgens. Meanwhile, in the hypogonadal group, LH displayed weak correlations with other markers, suggesting reduced feedback responsiveness or a disrupted axis. The onset age is positively correlated with SHBG. Conversely, the severity is negatively correlated with the total testosterone and free testosterone. Both results are interesting as they show ways to delay the onset age of hypogonadism and reduce its severity through appropriate hormonal treatments.
Psychosocially, gonadal dysfunction has a tangible impact on quality of life. Reports of erectile dysfunction and relational difficulties among hypogonadal males illustrate the broader implications beyond biochemical markers. Although current guidelines, such as the PNDS and DIDMOAD, provide valuable frameworks [
1,
3], none include systematic gonadal evaluation, despite its significant physical, emotional, and reproductive consequences.
Finally, given WS’s genetic basis, it is imperative to implement early screening and genetic counseling protocols. Hormonal assessment (total and free testosterone, LH, FSH, inhibin B) and semen analysis should begin by age 14 in males, particularly if puberty is delayed or incomplete. In females, early AMH evaluation, even in the context of apparently normal menstrual cycles, may uncover early ovarian insufficiency [
19,
20]. Fertility preservation strategies and psychological support must also be considered as part of comprehensive care. To date, only one article has specifically addressed genetic counseling in WS, led by the principal investigator of this study [
23], emphasizing the urgency of advancing research and clinical guidance in this area.
5. Conclusions
This study confirms that gonadal dysfunction is a prevalent and clinically significant feature of Wolfram syndrome (WS), predominantly manifesting as hypergonadotropic hypogonadism. Among males, 19 of 25 patients (76.0%) met diagnostic criteria for hypogonadism, including 12 cases of azoospermia and 2 cases of oligozoospermia. The mean total testosterone level in affected males was 6.36 ng/dL, substantially lower than the 18.08 ng/dL observed in the non-hypogonadal group. In females, 20% experienced premature menopause, and four individuals had AMH levels ≤ 0.5 ng/mL, with ovarian atrophy identified in three of these cases. These findings support primary gonadal failure as the predominant mechanism, although two patients exhibited hypogonadotropic hypogonadism, suggesting hypothalamic–pituitary axis involvement. Both cases of hypogonadotropic hypogonadism occurred in patients with early-onset insulin-dependent diabetes mellitus, as typically seen in WS. However, at the time of hormonal assessment, neither patient exhibited clinical signs of poor glycemic control (e.g., sustained hyperglycemia or ketoacidosis), nor were there indications of confounding factors such as significant weight loss, hyperprolactinemia, chronic illness, or neurological disease. These observations support the hypothesis that hypogonadotropic hypogonadism in these cases is likely intrinsic to the WS spectrum, rather than a secondary or functional phenomenon.
The high frequency of gonadal impairment—especially primary testicular failure in males and diminished ovarian reserve in females—highlights the urgent need to implement standardized protocols for gonadal function evaluation. Hormonal assessment (including testosterone, LH, FSH, and inhibin B) and semen analysis in boys from age 14, as well as AMH measurement and pelvic imaging in girls during early adolescence, should be incorporated into routine care. Given the profound psychosocial and reproductive consequences of hypogonadism, comprehensive care models must also include psychological support and tailored reproductive counseling. Furthermore, the hereditary nature of WS underscores the importance of structured genetic counseling. Future research should prioritize fertility preservation strategies to optimize long-term reproductive health and improve quality of life for individuals with WS.
Author Contributions
Conceptualization, G.E.-B.; methodology, G.E.-B.; software, J.L.F.-M.; validation, G.E.-B. and J.L.F.-M.; formal analysis, G.E.-B. and J.L.F.-M.; investigation, G.E.-B. and J.L.F.-M.; resources, G.E.-B.; data curation, G.E.-B. and J.L.F.-M.; writing—original draft preparation G.E.-B. and J.L.F.-M.; writing—review and editing, G.E.-B. and J.L.F.-M.; visualization, G.E.-B. and J.L.F.-M.; supervision, G.E.-B. and J.L.F.-M.; project administration, G.E.-B.; funding acquisition, G.E.-B. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the Andalusian Regional Ministry of Health (Consejería de Salud de Andalucía) for supporting this project through the AP-0009-2020-C1-F2 FPS 2020-Internal Study Code: 2/2021—Research Call for Primary Care, Regional Hospitals, and CHARES. Additionally, we thank the Consejería de Salud de Andalucía for its support through the “SAS 2024—Refuerzo con Recursos Humanos de la Actividad Investigadora en las Unidades Clínicas del Servicio Andaluz de Salud”; which has provided dedicated time for scientific investigation and contributed to the development of this study.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Torrecárdenas University Hospital (code 75/2020), approval date 27 February 2020.
Informed Consent Statement
Informed consent was obtained from all subjects (or their guardians) involved in the study.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We sincerely thank the families affected by Wolfram syndrome for their invaluable participation in this study, as well as the Spanish Association for Research and Support for Wolfram Syndrome for their continuous support and collaboration. We would like to acknowledge the work of the Spanish Wolfram Syndrome Clinical Team, which has been evaluating patients since 2011. We highlight the contributions of the specialties involved in this article, urology and endocrinology, with the participation of José Ignacio Abad Vivas-Pérez and Manuel Romero Muñoz, who have played a key role in patient assessment. Additionally, we recognize the incorporation of Pedro Mezquita Raya in recent years, further strengthening the team and contributing to the specialized evaluation of these clinical aspects.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Esteban-Bueno, G.; Romero Muñoz, M.; Navarro Cabrero, M.; Abad Vivas-Pérez, J.I.; Ekiza Bazán, J.; Belmonte García, T.; Ruiz-Castañeda, D.; Segura Luján, M. Clinical Practice Guideline for Wolfram Syndrome (DIDMOAD); CEASGA-Publishing: Soria, Spain, 2020; Available online: https://reunir.unir.net/handle/123456789/15796 (accessed on 8 April 2025).
- National Organization for Rare Disorders. Wolfram Syndrome: Symptoms, Causes, Treatment; NORD: Danbury, CT, USA, 2024; [Updated 2025 Mar; Cited 2025 Apr 8]; Available online: https://rarediseases.org/rare-diseases/wolfram-syndrome/ (accessed on 8 April 2025).
- Centre de Reference for Rare Eye Diseases (OPHTARA). National Protocol for Diagnosis and Care (PNDS): Wolfram Syndrome; Haute Autorité de Santé: Paris, France, 2019; Available online: https://www.has-sante.fr/jcms/p_3113807/fr/syndrome-de-wolfram-pnds (accessed on 8 April 2025).
- Esteban-Bueno, G.; Ruiz-Castañeda, D.; Ruiz-Martínez, J.; Romero-Muñoz, M.; Carrillo-Alascio, P. Natural history and clinical characteristics of 50 patients with Wolfram syndrome. Endocrine 2018, 61, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Rai, A.; Mavuduru, R.; Vaiphei, K.; Sharma, A.; Gupta, V.; Bhadada, S.K.; Lodha, S.; Panda, N.; Bhansali, A.; et al. Wolfram syndrome: Clinical and genetic profiling of a cohort from a tertiary care centre with characterization of the primary gonadal failure. Endocrine 2020, 69, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.G.; Bundey, S.E. Wolfram (DIDMOAD) syndrome. J. Med. Genet. 1997, 34, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Bueno, G.; Berenguel Hernández, A.M.; Fernández Fernández, N.; Navarro Cabrero, M.; Coca, J.R. Neurosensory Affectation in Patients Affected by Wolfram Syndrome: Descriptive and Longitudinal Analysis. Healthcare 2023, 11, 1888. [Google Scholar] [CrossRef]
- Esteban, G.; Ruano, M.; Motero, I. The quality of life in the relatives of Wolfram’s syndrome patients. Eur. J. Investig. Health Psychol. Educ. 2015, 5, 89–97. [Google Scholar] [CrossRef]
- Frontino, G.; Di Tonno, R.; Stancampiano, M.R.; Arrigoni, F.; Rigamonti, A.; Morotti, E.; Canarutto, D.; Bonfanti, R.; Russo, G.; Barera, G.; et al. Paediatric Wolfram syndrome type 1: Should gonadal dysfunction be considered a major criterion? Front. Endocrinol. 2023, 14, 1155644. [Google Scholar] [CrossRef]
- Jodoin, A.; Marchand, M.; Beltrand, J. Wolfram syndrome in a young woman with associated hypergonadotropic hypogonadism: A case report. J. Pediatr. Endocrinol. Metab. 2022, 35, 1552–1555. [Google Scholar] [CrossRef]
- Simsek, E.; Simsek, T.; Tekgül, S.; Hosal, S.; Seyrantepe, V.; Aktan, G. Wolfram (DIDMOAD) syndrome: A multidisciplinary clinical study in nine Turkish patients and review of the literature. Acta Paediatr. 2003, 92, 55–61. [Google Scholar] [CrossRef]
- Calapkulu, M.; Erkam, S.M.; Ozturk, U.I.; Duger, H.; Kizilgul, M.; Bostan, H.; Cakal, E.; Ozbek, M. A case of Wolfram syndrome with primary hypogonadism. Endocr. Abstr. 2020, 70, AEP601. [Google Scholar] [CrossRef]
- Salzano, G.; Rigoli, L.; Valenzise, M.; Chimenz, R.; Passanisi, S.; Lombardo, F. Clinical peculiarities in a cohort of patients with Wolfram syndrome 1. Int. J. Environ. Res. Public Health 2022, 19, 520. [Google Scholar] [CrossRef]
- Bonomi, M.; Vezzoli, V.; Krausz, C. Genetic architecture of congenital hypogonadotropic hypogonadism. Hum. Reprod. Open 2024, 2024, hoae053. [Google Scholar] [CrossRef]
- Bhasin, S.; Brito, J.P.; Cunningham, G.R.; Hayes, F.J.; Hodis, H.N.; Matsumoto, A.M.; Snyder, P.J.; Swerdloff, R.S.; Wu, F.C.; Yialamas, M.A. Testosterone therapy in men with hypogonadism: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2018, 103, 1715–1744. [Google Scholar] [CrossRef] [PubMed]
- Basaria, S. Male hypogonadism. Lancet 2014, 383, 1250–1263. [Google Scholar] [CrossRef]
- Ramasamy, R.; Armstrong, J.M.; Lipshultz, L.I. Preserving fertility in the hypogonadal patient: An update. Asian J. Androl. 2015, 17, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Spaziani, M.; Carlomagno, F.; Tarantino, C.; Angelini, F.; Vincenzi, L.; Gianfrilli, D. New perspectives in functional hypogonadotropic hypogonadism: Beyond late onset hypogonadism. Front. Endocrinol. 2023, 14, 1184530. [Google Scholar] [CrossRef]
- Dewailly, D.; Andersen, C.Y.; Balen, A.; Broekmans, F.; Dilaver, N.; Fanchin, R.; Griesinger, G.; Kelsey, T.W.; La Marca, A.; Lambalk, C.; et al. The physiology and clinical utility of anti-Mullerian hormone in women. Hum. Reprod. Update 2014, 20, 370–385. [Google Scholar] [CrossRef]
- Rigoli, L.; Bramanti, P.; Di Bella, C.; De Luca, F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr. Res. 2018, 83, 921–929. [Google Scholar] [CrossRef]
- Newell, L.; Cunningham, O.; Williams, D.; Barrett, T.; Dias, R. Hypogonadism and pubertal disorders in Wolfram syndrome. Endocr. Abstr. 2022, 85, OC7.3. [Google Scholar] [CrossRef]
- Rey, R.A.; Grinspon, R.P. Abnormalities of pubertal development and gonadal function in Noonan syndrome. Front. Endocrinol. 2023, 14, 1213098. [Google Scholar] [CrossRef]
- Esteban-Bueno, G.; Díaz-Anadón, L.R.; González, R.; Cabrero, N.; AM, B.H. Genetic protocol in Primary Care for rare diseases: Wolfram syndrome as a prototype. Aten. Primaria 2022, 54, 102280. [Google Scholar] [CrossRef]
- Toppings, N.B.; McMillan, J.M.; Au, P.Y.B.; Suchowersky, O.; Donovan, L.E. Wolfram syndrome: A case report and review of clinical manifestations, genetics, pathophysiology, and potential therapies. Case Rep. Endocrinol. 2018, 2018, 9412676. [Google Scholar] [CrossRef]
- Munari, E.V.; Amer, M.; Amodeo, A.; Bollino, R.; Federici, S.; Goggi, G.; Giovanelli, L.; Persani, L.; Cangiano, B.; Bonomi, M. The complications of male hypogonadism: Is it just a matter of low testosterone? Front. Endocrinol. 2023, 14, 1201313. [Google Scholar] [CrossRef] [PubMed]
- Boehm, U.; Bouloux, P.-M.; Dattani, M.T.; de Roux, N.; Dodé, C.; Dunkel, L.; Dwyer, A.A.; Giacobini, P.; Hardelin, J.-P.; Juul, A.; et al. European Consensus Statement on congenital hypogonadotropic hypogonadism—Pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharefi, A.; Quinton, R. Current national and international guidelines for the management of male hypogonadism: Helping clinicians to navigate variation in diagnostic criteria and treatment recommendations. Endocrinol. Metab. 2020, 35, 526–540. [Google Scholar] [CrossRef]
- Rohayem, J.; Cunningham, O.; Williams, D.; Wistuba, J.; McCarthy, L.; Barrett, T.G.; Dias, R.P. Gonadal function in males with WFS1 spectrum disorder (Wolfram syndrome)—A European cohort perspective. Andrology 2025. Epub ahead of print. [Google Scholar] [CrossRef]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).








{kind=link}
{kind=link}
{kind=link}
{kind=link}