
The Clinical and Laboratory Profiles of a Deletional α2-Globin Gene Polyadenylation Signal Sequence (AATAAA > AATA--) [HBA2:c.*93_*94delAA]: The Malaysian Experience

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Mutation Analysis
2.2. Statistical Analysis
2.3. Ethics Approval
3. Results
3.1. Demographic Profile
3.2. Genotyping Group
3.3. Haematological Characteristics and Clinical Phenotype
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harteveld, C.L.; Higgs, D.R. Alpha-thalassaemia. Orphanet. J. Rare Dis. 2010, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Kousiappa, I.; Papasavva, T.; Christopoulos, G.; Pavlou, E.; Petrou, M.; Feleki, X.; Karitzie, E.; Phylactides, M.; Fanis, P.; et al. The molecular spectrum and distribution of haemoglobinopathies in Cyprus: A 20-year retrospective study. Sci. Rep. 2016, 6, 26371. [Google Scholar] [CrossRef]
- Amid, A.; Lal, A.; Coates, T.D.; Fucharoen, S. (Eds.) Guidelines for the Management of α-Thalassaemia; Thalassaemia International Federation: Nicosia, Cyprus, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK602223/ (accessed on 20 February 2025).
- Goh, L.P.W.; Chong, E.T.J.; Lee, P.C. Prevalence of Alpha(α)-Thalassemia in Southeast Asia (2010–2020): A Meta-Analysis Involving 83,674 Subjects. Int. J. Env. Res. Public Health 2020, 17, 7354. [Google Scholar] [CrossRef] [PubMed]
- Farashi, S.; Harteveld, C.L. Molecular basis of α-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Nainggolan, I.M.; Harahap, A.; Setianingsih, I. Hydrops fetalis associated with homozygosity for Hb Adana [α59(E8)Gly→Asp (α2)]. Hemoglobin 2010, 34, 394–401. [Google Scholar] [CrossRef]
- Lee, T.; Lai, M.; Ismail, P.; Ramachandran, V.; Tan, J.; Teh, L.; Othman, R.; Hussein, N.; George, E. Analysis of α1 and α2 globin genes among patients with hemoglobin Adana in Malaysia. Genet. Mol. Res. 2016, 15, gmr.15027400. [Google Scholar] [CrossRef]
- Ahmad, R.; Saleem, M.; Aloysious, N.S.; Yelumalai, P.; Mohamed, N.; Hassan, S. Distribution of alpha thalassaemia gene variants in diverse ethnic populations in malaysia: Data from the institute for medical research. Int. J. Mol. Sci. 2013, 14, 18599–18614. [Google Scholar] [CrossRef]
- Idris, F.; Liew, C.Y.; Seman, Z.; Mahmud, N. Optimal Mean Corpuscular Haemoglobin (MCH) Cut-Off Value for Differentiating Alpha Plus and Alpha Zero Thalassaemia in Thalassaemia Screening. Mal. J. Med. Health Sci. 2020, 2004, 69–74. [Google Scholar]
- Yasin, N.M.; Musa, N.H.; Hamid, F.S.; Hassan, S.; Aziz, N.A.; Sahid, E.N.; Yusoff, Y.M.; Jaafar, A.A.; Esa, E. Haematological and Molecular Characteristics of Hb Singapore [HBA2:c. 425G > C] Unique Among the Malays from Kelantan, Malaysia. Malays. J. Human. Genet. 2022, 3, 1–8. [Google Scholar]
- Yasin, N.M. A Rare Interactions Between Heterozygous --SEA Deletion and Hb Ube-2 [α68 (E17) Asn →Asp]; First Reported Case from Malaysia. Biomed. J. Sci. Tech. Res. 2023, 51, 42867–42869. [Google Scholar] [CrossRef]
- Harteveld, C.L.; Losekoot, M.; Haak, H.; Heister, G.A.; Giordano, P.C.; Bernini, L.F. A novel polyadenylation signal mutation in the alpha 2-globin gene causing alpha thalassaemia. Br. J. Haematol. 1994, 87, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Farashi, S.; Garous, N.F.; Ashki, M.; Vakili, S.; Zeinali, F.; Imanian, H.; Azarkeivan, A.; Giordano, P.C.; Najmabadi, H. Homozygosity for the AATAAA > AATA- - Polyadenylation Site Mutation on the α2-Globin Gene Causing Transfusion-Dependent Hb H Disease in an Iranian Patient: A Case Report. Hemoglobin 2015, 39, 355–358. [Google Scholar] [PubMed]
- Kalle Kwaifa, I.; Lai, M.I.; Md Noor, S. Non-deletional alpha thalassaemia: A review. Orphanet J. Rare Dis. 2020, 15, 166. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L.; Wallace, R.B.; Pressley, L.; Clegg, J.B.; Weatherall, D.J.; Higgs, D.R. The polyadenylation site mutation in the alpha-globin gene cluster. Blood 1988, 71, 313–319. [Google Scholar] [CrossRef]
- Haider, M.; Adekile, A. Alpha-2-globin gene polyadenylation (AATAAA-->AATAAG) mutation in hemoglobin H disease among Kuwaitis. Med. Princ. Pract. 2005, 14 (Suppl. S1), 73–76. [Google Scholar] [CrossRef]
- Deshpande, P.; Kamalanathan, N.; Sampath, E.; George, B.; Shaji, R.V.; Edison, E.S. Characterization of Clinical and Laboratory Profiles of the Deletional α2-Globin Gene Polyadenylation Signal Sequence (AATAAA > AATA- -) in an Indian Population. Hemoglobin 2015, 39, 415–418. [Google Scholar] [CrossRef]
- Yüregir, G.T.; Aksoy, K.; Cürük, M.A.; Dikmen, N.; Fei, Y.J.; Baysal, E.; Huisman, T.H. Hb H disease in a Turkish family resulting from the interaction of a deletional alpha-thalassaemia-1 and a newly discovered poly A mutation. Br. J. Haematol. 1992, 80, 527–532. [Google Scholar] [CrossRef]
- Harteveld, C.L.; Oosterhuis, W.P.; Schoenmakers, C.H.; Ananta, H.; Kos, S.; Bakker Verweij, M.; van Delft, P.; Arkesteijn, S.G.; Phylipsen, M.; Giordano, P.C. Alpha-thalassaemia masked by beta gene defects and a new polyadenylation site mutation on the alpha2-globin gene. Eur. J. Haematol. 2010, 84, 354–358. [Google Scholar] [CrossRef]
- Ren, Z.M.; Li, W.J.; Xing, Z.H.; Fu, X.Y.; Zhang, J.Y.; Chen, Y.S.; Li, D.F. Detecting rare thalassemia in children with anemia using third-generation sequencing. Hematology 2023, 28, 2241226. [Google Scholar] [CrossRef]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood 2000, 95, 360–362. [Google Scholar] [CrossRef]
- Eng, B.; Patterson, M.; Walker, L.; Chui, D.H.; Waye, J.S. Detection of severe nondeletional alpha-thalassemia mutations using a single-tube multiplex ARMS assay. Genet. Test. 2001, 5, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.W.; Higgs, D.R.; Murphy, P.; Villegas, A.; de Miguel, A. A mutation in the polyadenylation signal of the alpha 2 globin gene (AATAAA-->AATA--) as a cause of alpha thalassaemia in Asian indians. Br. J. Haematol. 1994, 88, 225–227. [Google Scholar] [CrossRef]
- Laosombat, V.; Fucharoen, S.; Wiriyasateinkul, A. Interaction of the α2 polyadenylation signal mutation (AATAAA → AATA– –) and α0-THALASSEMIA (– –SEA), resulting in Hb H disease in a Thai patient. Hemoglobin 2001, 25, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An Interactive Database for Haemoglobin Variants and Epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef]
- Prior, J.F.; Lim, E.; Lingam, N.; Raven, J.L.; Finlayson, J. A Moderately Severe α-Thalassemia Condition Resulting From a Combination of the α2 Polyadenylation Signal (AATAAA→AATA– –) Mutation and a 3.7 Kb α Gene Deletion in an Australian Family. Hemoglobin 2007, 31, 173–177. [Google Scholar] [CrossRef]
- Henderson, S.; Chapple, M.; Rugless, M.; Fisher, C.; Kinsey, S.; Old, J. Haemoglobin H hydrops fetalis syndrome associated with homozygosity for the alpha2-globin gene polyadenylation signal mutation AATAAA-->AATA- -. Br. J. Haematol. 2006, 135, 743–745, Erratum in Br. J. Haematol. 2007, 136, 680. [Google Scholar] [CrossRef]
- Azma, R.Z.; Ainoon, O.; Hafiza, A.; Azlin, I.; Noor Farisah, A.R.; Nor Hidayati, S.; Noor Hamidah, H. Molecular characteristic of alpha thalassaemia among patients diagnosed in UKM Medical Centre. Malays. J. Pathol. 2014, 36, 27–32. [Google Scholar] [PubMed]
- Rahimah, A.N.; Nisha, S.; Safiah, B.; Roshida, H.; Punithawathy, Y.; Nurul, H.; Syahzuwan, H.; Zubaidah, Z. Distribution of alpha thalassaemia in 16 year old Malaysian Students in Penang, Melaka and Sabah. Med. J. Malaysia. 2012, 67, 565–570. [Google Scholar] [PubMed]
- Yasin, N.M.; Abdul Hamid, F.S.; Hassan, S.; Mat Yusoff, Y.; Mohd Sahid, E.N.; Esa, E. Characterization of New Alpha Zero (α0) Thalassaemia Deletion (--GB) among Malays in Malaysian Population. Diagnostics 2023, 13, 3286. [Google Scholar] [CrossRef]
- Curinha, A.; Braz, S.O.; Pereira-Castro, I.; Cruz, A.; Moreira, A. Implications of polyadenylation in health and disease. Nucleus 2014, 5, 508–519. [Google Scholar] [CrossRef]
- Clerici, M.; Faini, M.; Muckenfuss, L.M.; Aebersold, R.; Jinek, M. Structural basis of AAUAAA polyadenylation signal recognition by the human CPSF complex. Nat. Struct. Mol. Biol. 2018, 25, 355. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, T.P.; Smetanina, N.S.; Huisman, T.H. A second, elongated, alpha 2-globin mRNA is present in reticulocytes from normal persons and subjects with terminating codon or poly A mutations. Biochem. Biophys. Res. Commun. 1995, 214, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
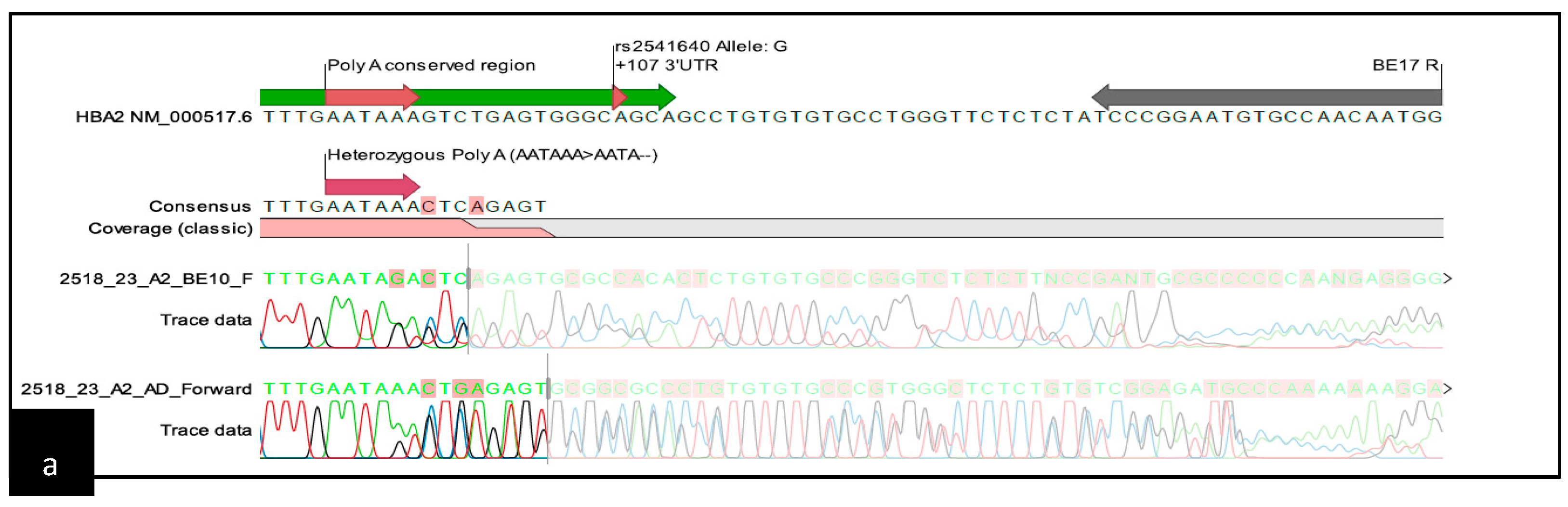
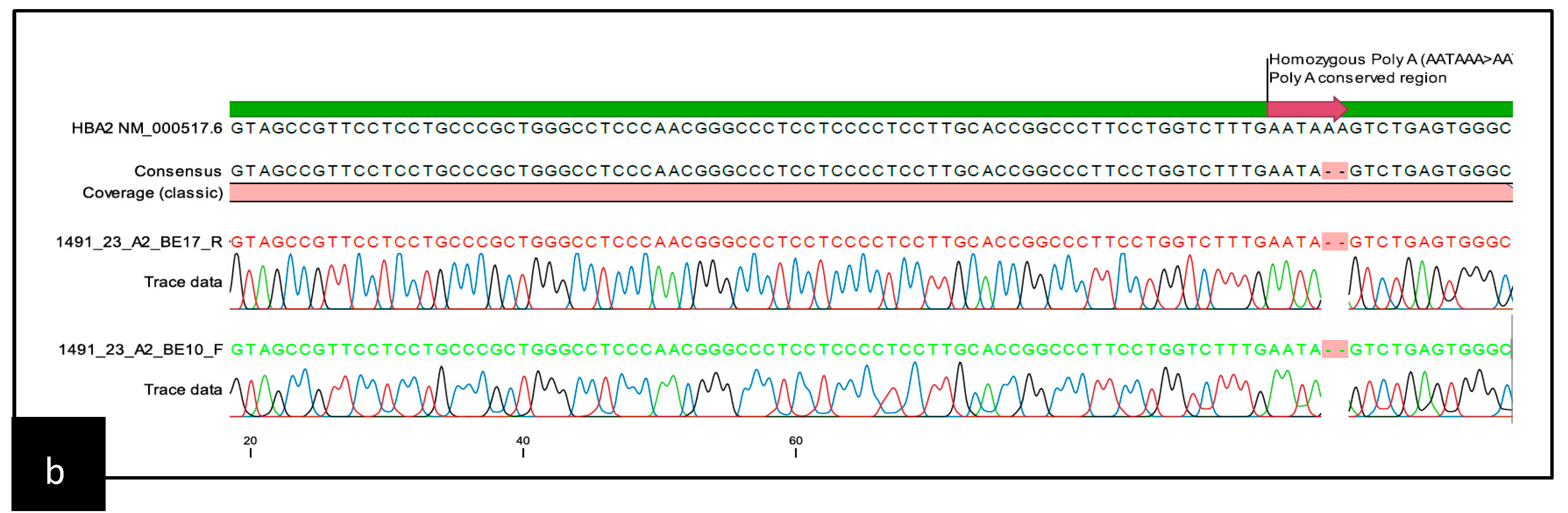


{kind=link}
{kind=link}
{kind=link}
| Genotype | α−AAα/αα | α−AAα/α−AAα | α−AAα/−α3.7 | α−AAα/--SEA | α−AAα/αCSα | α−AAα/αAdanaα | α−AAα/αPakseα, βEβ |
|---|---|---|---|---|---|---|---|
| n (%) | 22 | 1 | 2 | 2 | 3 | 1 | 1 |
| RBC (106/uL) | 5.5 ± 0.52 | 4.0 | 5.9 | 3.8 | 4.7 ± 0.29 | 3.1 | 4.8 |
| Hb (g/dL) | 12.4 ± 1.42 | 8.0 | 11.1 ± 1.90 | 8.3 ± 1.27 | 8.9 ± 0.61 | 5.6 | 8.6 |
| MCV (fL) | 69.8 ± 3.07 | 70.0 | 58.4 ± 4.94 | 75.3 ± 2.19 | 66.0 ± 2.75 | 91.0 | 59.3 |
| MCH (pg) | 22.2 ± 1.57 | 20.0 | 18.5 ± 0.98 | 22.1 ± 2.96 | 18.8 ± 0.20 | 28.0 | 18.0 |
| RDW-CV (%) | 15.38 ± 1.08 | 38.5 | 18.4 ± 0.14 | 21.3 | 22.3 ± 3.34 | 27.7 | 19.4 |
| CE (%) | |||||||
| Hb A | 96.5 ± 3.37 | 75.8 | 97.7 | 79.6 | 96.0 ± 2.36 | 82.1 | |
| Hb A2 | 2.4 ± 0.30 | 1.1 | 2.3 | 1.9 | 1.5 ± 0.3 | 2.3 | |
| Hb F | 0.3 ± 0.00 | 1.1 | |||||
| Hb variant | NA | Z15 (23.1%) | NA | Z15 (18.5%) | ** CS (2.1), Z12 (2.7) and Z15 (0.6) | E, 14.5 | |
| HPLC (%) | |||||||
| Hb A | 86.0 ± 0.9 | 87.0 | 87.2 ± 1.77 | ||||
| Hb A2 | 2.6 ± 0.10 | 1.6 | 2.3 | 1.8 | 2.3 ± 0.10 | 2.5 | |
| Hb F | 0.6 ± 0.72 | 0.0 | 1.8 | 0.5 | 0.8 ± 0.78 | 0.5 | |
| Hb Variant | NA | Pre-run peak | Pre-run peak | Pre-run peak, CS (1.5) | Pre-run peak | ||
| Gel electrophoresis pH8.5 | NA | Fast band at H region | Fast band at H region | No H band | |||
| H-Inclusion | NA | Positive | Many inclusions seen | Negative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasin, N.M.; Hassan, S.; Aziz, N.A.; Abdul Hamid, F.S.; Esa, E.; Zulkefli, E.S.; Ghazali, R.; Tajuddin, S.N.; Darawi, M.N.; Yusoff, Y.M.; et al. The Clinical and Laboratory Profiles of a Deletional α2-Globin Gene Polyadenylation Signal Sequence (AATAAA > AATA--) [HBA2:c.*93_*94delAA]: The Malaysian Experience. Diagnostics 2025, 15, 1284. https://doi.org/10.3390/diagnostics15101284
Yasin NM, Hassan S, Aziz NA, Abdul Hamid FS, Esa E, Zulkefli ES, Ghazali R, Tajuddin SN, Darawi MN, Yusoff YM, et al. The Clinical and Laboratory Profiles of a Deletional α2-Globin Gene Polyadenylation Signal Sequence (AATAAA > AATA--) [HBA2:c.*93_*94delAA]: The Malaysian Experience. Diagnostics. 2025; 15(10):1284. https://doi.org/10.3390/diagnostics15101284
Chicago/Turabian StyleYasin, Norafiza Mohd, Syahzuwan Hassan, Nur Aisyah Aziz, Faidatul Syazlin Abdul Hamid, Ezalia Esa, Ezzanie Suffya Zulkefli, Rohana Ghazali, Syirah Nazirah Tajuddin, Mohd Nazif Darawi, Yuslina Mat Yusoff, and et al. 2025. "The Clinical and Laboratory Profiles of a Deletional α2-Globin Gene Polyadenylation Signal Sequence (AATAAA > AATA--) [HBA2:c.*93_*94delAA]: The Malaysian Experience" Diagnostics 15, no. 10: 1284. https://doi.org/10.3390/diagnostics15101284
APA StyleYasin, N. M., Hassan, S., Aziz, N. A., Abdul Hamid, F. S., Esa, E., Zulkefli, E. S., Ghazali, R., Tajuddin, S. N., Darawi, M. N., Yusoff, Y. M., & Harteveld, C. L. (2025). The Clinical and Laboratory Profiles of a Deletional α2-Globin Gene Polyadenylation Signal Sequence (AATAAA > AATA--) [HBA2:c.*93_*94delAA]: The Malaysian Experience. Diagnostics, 15(10), 1284. https://doi.org/10.3390/diagnostics15101284