1. Introduction
Idiopathic thrombocytopenic purpura (ITP) is an autoimmune disease defined by the destruction of platelets caused by the effect primary antiplatelet antibodies have against platelet glycoproteins (GPIIb/IIIa and GPIb/IX). There are three categories of ITP: (1) newly diagnosed idiopathic thrombocytopenic purpura (ndITP) (within 3 months from diagnosis), (2) persistent ITP (from 3 to 12 months) and (3) chronic idiopathic thrombocytopenic purpura (chITP) (lasting for more than 12 months) [
1].
From 10 to 30% of patients with ndITP have thrombocytopenia longer than 12 months, which develops into chITP [
2]. Factors at presentation that can predict which patients will develop the disease have not been defined. It is difficult to predict not only the length of achieved remission (both clinical or laboratory) in ndITP and chITP, but also which 20% of patients with chITP will spontaneously recover [
2,
3,
4].
Previously, it was considered that chITP rarely occurs in childhood, and that children with chITP are more likely to attain spontaneous remission compared to adult patients [
5]. chITP in children most commonly develops as the continuation of acute and persistent ITP. Patients’ condition deteriorates following viral infections or the intake of non-steroidal anti-rheumatic drugs [
5,
6]. More serious signs of hemorrhagic syndrome are shown in children, whereas higher comorbidity rates are found in adults [
5,
6,
7]. Clinical expressions of ITP are extremely diverse, ranging from asymptomatic to life-threatening hemorrhages [
1,
7,
8].
ITP is a diagnosis of exclusion; however, in a large cohort of nearly 4000 pediatric patients with chITP included in the registry, misdiagnosis was confirmed in approximately 3% of cases, predominantly as secondary ITP associated with underlying conditions [
1,
9]. It is presumed that other forms of ITP, such as those occurring in the context of systemic lupus erythematosus, antiphospholipid syndrome, connective tissue disorders, or neoplastic diseases, are similarly characterized by the binding of antiplatelet autoantibodies to specific glycoproteins or phospholipids on platelet surfaces. To the authors’ knowledge, no studies to date have investigated the emergence of other autoimmune diseases during the course of chITP; rather, existing studies have focused solely on whether an alternative diagnosis may have been masked by the initial diagnosis of chITP. Furthermore, data from the registry indicate that only 1–2% of ITP patients have a positive family history, thereby raising the question of whether ITP may have a genetic component [
10].
According to recommendations by multiple authors, treatment is required for children with chITP with signs of hemorrhagic syndrome, regardless of the platelet count. Their recommendations are contradictory for patients with chITP who present with very low platelet counts (less than 10–20 × 10
9/L) and no bleeding manifestations. On the other hand, treatment is not required for asymptomatic patients with chITP and moderate thrombocytopenia (about 50 × 10
9/L). These patients may present with bleeding manifestations during surgical interventions, substantial injuries, or viral infections. It is considered that in patients with chITP, stable platelet counts are measured for years [
1,
7,
8,
11,
12]. Approximately 60–70% of patients with chITP require some form of treatment, while others may experience spontaneous recovery. However, the factors that predict which patients will recover spontaneously have not yet been defined [
1,
2,
3,
4].
Data from the literature suggest that around 25% of patients with chITP failed to respond to standard initial first-line therapy (intravenous immunoglobulins, systemic corticosteroids) and to splenectomy [
7,
8,
11,
12]. In the last decade, there has been an increase in the clinical use of thrombopoietin receptor agonists, and preliminary results are very encouraging for both acute and chronic cases [
13,
14]. Various cytotoxic and immunosuppressive drugs are introduced as second-line therapy, either alone or in combination with other drugs and dosages. The drugs are experimentally administered, and their exact mechanism of action in ITP is not very clear [
1,
7,
8,
11,
12]. Regardless of the number of cases, 5 to 10% of children with chITP are unresponsive to administered therapy. There is no consensus concerning the treatment of these patients which represents a serious challenge for clinicians. Increasingly, the term “refractory ITP” is being introduced to describe such cases [
1,
4,
6,
7,
8,
11,
12,
13,
14,
15].
The aim of this study is, based on the data from a representative number of cases, to present the most significant medical history and clinical and laboratory characteristics of children with diagnosed chITP.
3. Results
The total number of children diagnosed with chITP was 152, with a mild female preponderance (F:M = 85:67, i.e., 1.27:1). The average age of our patients was 12.3 ± 3.8 years. According to age, most children were adolescents (35%), with smaller percentages of school and preschool children (around 25% in both groups) and the smallest group comprising small children. It was determined that almost 70% of all patients who had some other autoimmune disease beside chITP were females. On the other hand, 70% of patients with chITP who had positive antinuclear antibodies (ANA) or lupus anticoagulant antibodies (LAC) were exclusively males.
Table 1 shows the case distribution of children with chITP in percentages according to the severity of bleeding manifestations based on the BSS, dominant bleeding manifestations, and the need for transfusion of deplasmatized erythrocytes and platelet concentrates for the duration of disease.
From all of the observed parameters, the BSS showed correlation with platelet count in inverse proportion. It was determined that patients who received transfusions had the same ABO blood system distribution as the general population. On the other hand, 40% of patients (17 of 45) who received transfusions were Rh negative, and all the patients who received transfusions had severe bleeding manifestations (BSS 3 and 4).
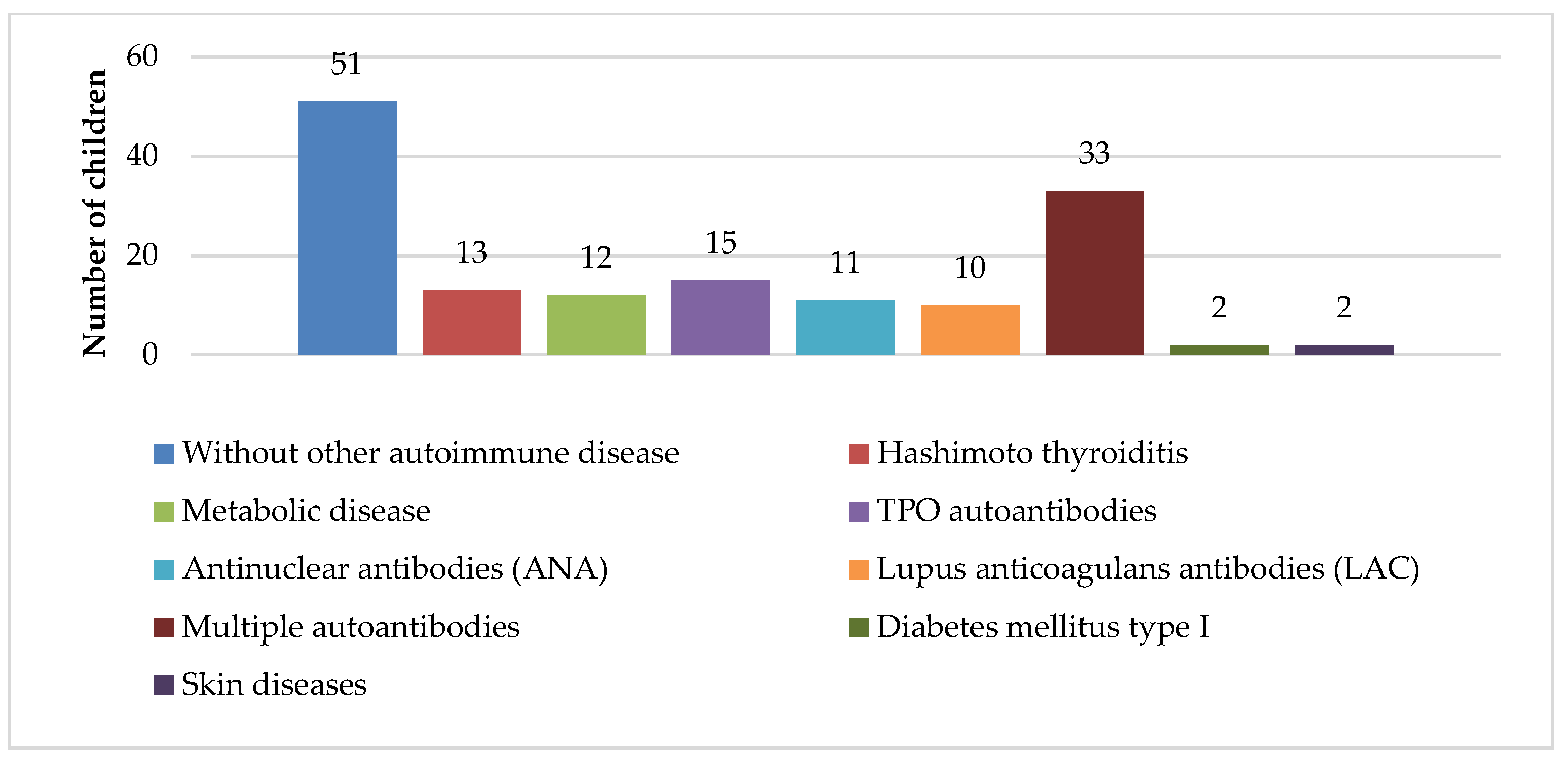
Figure 1 shows the distribution of other autoimmune diseases in children with chITP. Thirty-five percent of patients had chITP alone, whereas Hashimoto thyroiditis was the most frequent comorbidity found in 15% of patients, and high titer levels of autoantibodies were found in 45%, but without any clinical manifestation of the autoimmune diseases.
A statistically significant difference was noted regarding the duration of disease when the group of patients with chITP alone was compared with the group of patients that were diagnosed with an autoimmune disease. This is because a high percentage of children with autoimmune comorbidities had symptoms for more than 2 years, and most of them had severe bleeding manifestations of ITP (
Table 2).
In almost the same number of family members of children with chITP, there was no report of autoimmune diseases (65 of 152 patients), or more than one family member was diagnosed with an autoimmune disease (60 of 152). Patients with autoimmune comorbidities and chITP most commonly have a positive family medical history (
Table 3). Comparing the groups with positive and negative family medical histories showed that children with chITP who had relatives with autoimmune diseases were affected for more than 2 years (37 of 46) and were resistant to initial therapy (32 of 37).
Around 55% of patients with chITP had temporary response (<30 days) after initial therapy in newly diagnosed form, 25% were resistant ITP patients, whereas in 20% of patients, the response to initial therapy was good, lasting a few months or long term, mostly with complete clinical remission.
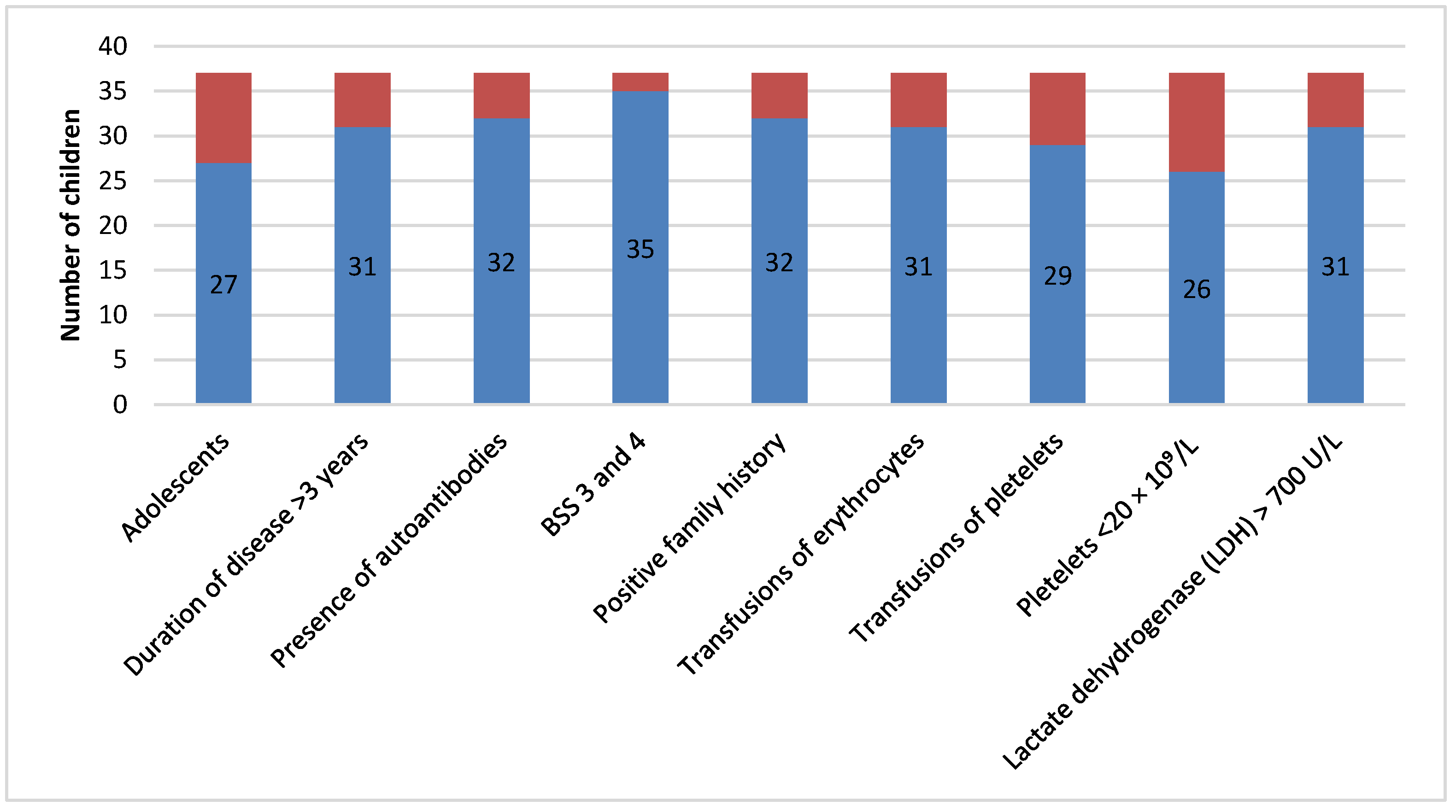
Figure 2 shows patients resistant to initial therapy who were most commonly adolescents. These patients presented with long duration of disease, severe bleeding, required many transfusion treatments, and had more comorbidities, a positive family medical history, lower platelet count, and higher LDH values. Furthermore, they were more resistant to corticosteroid therapy.
Stool samples from slightly more than 20% of patients tested positive for
Helicobacter pylori antigen (
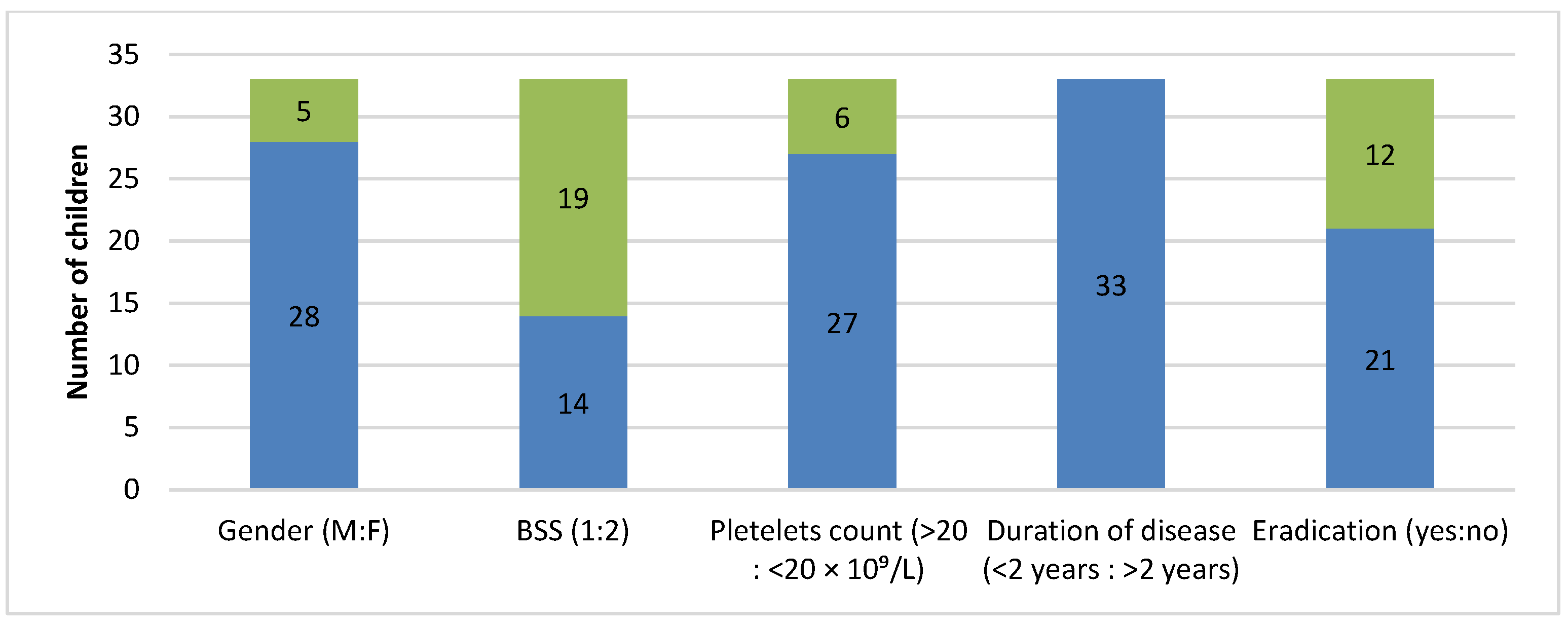
Figure 3). It was noticeable that these patients were mostly males, asymptomatic or with mild bleeding manifestations, had a platelet count of 20–50 × 10
9/L, and characterized by a short duration of disease (<2 years). The infection was completely eradicated in 21 patients.
Abdominal ultrasound detected splenomegaly in almost 25% of patients (37 of 152 patients), whereas the number of patients with splenomegaly was lower during the splenectomy (18 of 152). Moreover, it was noted that 14% of patients (21 of 152) had accessory spleen. It was determined that patients with splenomegaly had a platelet count lower than 20 × 109/L (32 of 37) and mean platelet volume (MPV) higher than 10 fL (36 of 37). No correlation with other examined parameters was noted.
Mean platelet count at diagnosis of chITP (around 12 months) was 35.7 ± 9.6 × 10
9/L, and MPV was 10.6 ± 1.4 fL. All the children with chITP underwent cytological examination of bone marrow aspirate, and no dysplastic changes were noted (
Table 4).
When comparing the three groups of patients with different platelet counts, statistically significant differences were noted in the duration and severity of disease, number of received transfusions, and resistance to initial therapy (
Table 5).
It was determined that all the patients (17 of 17) with extremely large platelets (MPV > 12 fL) who had a platelet count of <20 × 109/L, belonged to the group with mild bleeding manifestations (BSS 1 and 2) and had increased production of megakaryocytes in the bone marrow. On the other hand, all the patients (23 of 23) with normal values of MPV (7–10 fL), who had a platelet count <20 × 109/L, belonged to the group with severe bleeding manifestations (BSS 3 and 4) and had rare megakaryocytes in the bone marrow. This was not found in patients with mildly increased values of MPV (10–12 fL) and a platelet count >20 × 109/L. Furthermore, no correlation between MPV and other examined parameters was noted.
There was no correlation between the examined parameters and normal or increased production of megakaryocytes in the bone marrow of patients with chITP. However, all patients with rare megakaryocytes in the bone marrow (34 of 34) had some autoimmune disease or were positive for autoantibodies, and many (24 of 34) had severe bleeding manifestations, received more transfusions (23 of 34), and were resistant to initial therapy (26 of 34 patients).
Almost 70% of patients had higher levels of LDH, 450–1000 U/L, and in almost a third of these, the level ranged from 700 to 1000 U/L. It was observed that extremely high LDH values (more than 700 U/L) correlated with low platelet counts <20 × 109/L and resistance to initial therapy.
Only three patients with chITP had sufficient serum vitamin D levels ranging from 30 to 40 ng/mL. Around 25% of patients had hypovitaminosis D ranging from 20 to 30 ng/mL, whereas the others had vitamin D levels lower than 20 ng/mL, 17% of which had vitamin D insufficiency (<10 ng/mL). Despite the high percentage of children with chITP who had hypovitaminosis D, vitamin D insufficiency alone (<10 ng/mL) was correlated with a higher BSS (26 of 26 patients), whereas no correlation with other examined parameters was noted.
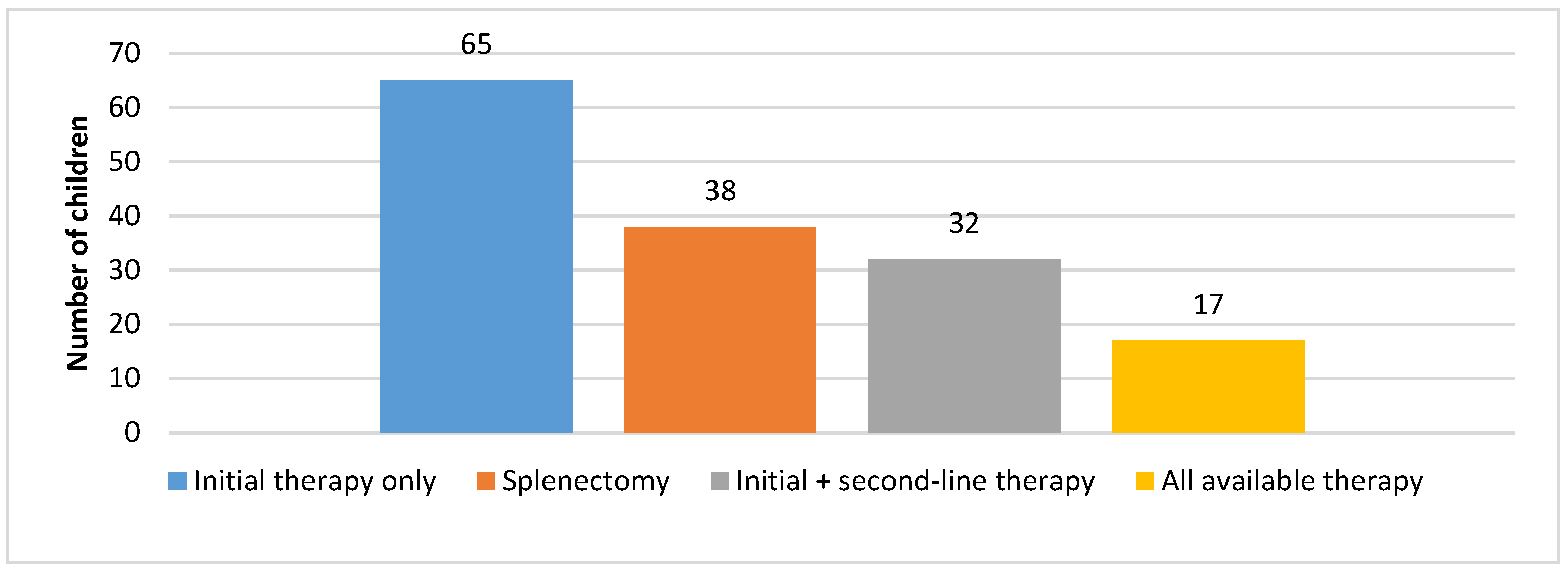
Figure 4 shows therapy administration in children with chITP. Approximately 45% of patients required only initial therapy in the first 12 months; afterwards, they entered a stable phase. Initial therapy and available second-line therapy were administered to 25% of patients. Around 20% of patients underwent splenectomy as the remaining treatment method, whereas 10% of patients received this treatment as part of second or third-line therapy.
4. Discussion
Our study of 152 cases of chITP showed that the ratio between sexes matched the data from the literature (F:M~1.3:1). Adolescents accounted for the highest percentage of affected patients (approximately 35%) [
1,
2,
7]. One of the most reported epidemiological differences between the populations of children and adults with ITP was certainly the preponderance of females among adult patients (~2:1 ratio), while in children, the ratio was 1.3:1. This was most certainly associated with the increased incidence of autoimmune diseases in adult female patients [
5,
6]. Moreover, almost 70% of all patients who had autoimmune diseases were females. Official data from the literature show that disease development into chITP is very common in female children older than 10 years of age; however, numerous studies show that this development is attributed to neither the sex nor age of the patients [
3,
4].
After analyzing the data from patients’ medical histories, the absence of a clear cause (such as previous infections, vaccinations, insect bites or stings, etc.) is one of the commonly mentioned markers in the literature that can point to chITP in patients with ndITP. However, this may not be the rule [
3,
4,
16]. In this cohort, at diagnosis, almost 65% of patients provided no data on a clear cause that initiated the immunological response.
In our study, approximately 15% of examined patients were asymptomatic, and 45% had skin hematomas, whereas 35% of patients had significant bleeding symptoms. Epistaxis and gingival bleeding are common accompanying symptoms and rarely dominant bleeding manifestations. To date, clinicians estimate the degree of bleeding. Although there have been attempts to establish a bleeding assessment scale [
17,
18], discrepancies among researchers in this field are still present. Most certainly, the percentage of asymptomatic patients with chITP ranges from 10 to 70% in different episodes [
3,
4,
6,
7,
19]. In this study, we regarded patients as asymptomatic with complete clinical remission, however, with persisting thrombocytopenia after their first acute bleeding episode. Moreover, in the second year of the duration of disease, almost 45% of patients had stable platelet counts and were asymptomatic. BSS was graded 2 in approximately 55% of patients.
Table 1 shows that only 15% of patients had severe, at times life-threatening, bleeding episodes (BSS of 4), which is consistent with data from the literature [
3,
4,
6,
7,
19].
According to the aforementioned data, transfusion of concentrated thrombocytes or deplasmatized erythrocytes was not required for approximately 70% of patients. On the other hand, 20% of patients had to be given more than 10 transfusion treatments for the duration of disease. Transfusion was required during certain bleeding episodes for the rest of the patients. This percentage of patients is much higher than the percentages cited in the results by other researchers (between 5 and 15%) [
1,
3,
4,
6,
7,
19]. However, it must be considered that our study included children from Serbia with chITP in the last 25 years, in a period of national economic, political, and healthcare transition. Therefore, potential unavailability of other therapy options presumably caused an increase in the number of patients who received transfusion. It was noted that patients showed an ABO blood system distribution that was no different from that in the general population. Moreover, it should be noted that approximately 40% of patients who received transfusions were Rh negative. To date, there are no cases in which this has been documented.
An interesting difference between pediatric patients and adult chronic patients with ITP is the incidence of comorbid conditions—the presence of ≥1 comorbidity in only 5% of pediatric patients and >30% of adults at presentation, whereas after 2 years of measurement, comorbidities were present in 10–15% of pediatric patients and in 35–50% of adults [
3,
4]. In our study, approximately 35% of patients were diagnosed with ITP alone, whereas Hashimoto thyroiditis was diagnosed as a comorbidity in only 15% of patients. Few patients were diagnosed with hormonal or metabolic imbalance (polycystic ovary syndrome, insulin resistance, obesity), type I diabetes, vitiligo, or psoriasis. The remaining 45% of patients had high titer levels of antithyroglobulin antibodies, ANA and/or LAC antibodies, alone or in combination; however, no clinical manifestations of the autoimmune disease were noted (
Figure 1). Positive test results for autoantibodies are mentioned in other studies; however, to date, their significance has not been fully understood [
1,
3,
4,
20,
21,
22]. While certain authors recommend monitoring and associate ANA positivity with response to therapy [
19], others question whether this is truly necessary [
21,
22]. A very interesting result was obtained in our study: when analyzing only patients with chITP and positive for antithyroglobulin antibodies, ANA, and/or LAC antibodies, either individually or in combination, but without clinical manifestations of autoimmune disease, it was found that nearly 70% of these patients were boys (47 out of 68), a finding that, to date, has not been documented in the literature. However, due to the small number of patients included in this and other studies, more extensive research is needed. The authors intend to analyze the outcomes of this specific subgroup of chITP patients positive for autoantibodies and publish the findings in a subsequent study. In our study, a higher percentage of children with chITP and autoimmune comorbidities were affected for more than 2 years (20% with no comorbidities compared to 35% with comorbidities), and a higher percentage of patients had severe bleeding manifestations (6% compared to 40%) (
Table 2). Furthermore, 50% of these patients had a positive family medical history of autoimmune diseases.
Approximately 45% of families of children with ITP reported no autoimmune diseases. However, autoimmune diseases were reported in more than one family member of the patients in almost the same percentage. Cases with a single reported disease in the family are rare, with functional disorder of the thyroid gland being the most common one. Only five patients had relatives who suffered from ITP. It is shown that a high percentage of children with chITP who had relatives with autoimmune diseases were affected for more than 2 years and resistant to initial therapy (
Table 3). Several authors tried to determine attribution of genetic predispositions and the importance of autoimmune diseases in the family history for the onset of ITP and development of chITP in their studies. The authors do agree that both the onset and development of ITP can be attributed to a positive family history. However, to date, there are no consistent results regarding genetic predisposition according to ITP development [
1,
2,
21].
Our patients with the newly diagnosed form of ITP did not go through the “wait and see” approach. Approximately 55% of the patients had a temporary response to initial therapy. Most commonly, it included laboratory, but rarely clinical, relapses that occurred 5–6 weeks following intravenous immunoglobulin treatment, lowering doses of Prednisone, or after no clinically meaningful response to pulse Dexamethasone was noted. Around 25% of patients were resistant to therapy (unachieved remissions or temporary relapse), whereas 20% had a good response to therapy that lasted for a few months or achieved complete clinical remission. Apart from the aforementioned standard therapy options for the treatment of ndITP, in the last decade, other approaches that include certain second-line therapies have emerged. However, the results obtained by researchers who examined the effects of standard therapy were similar to the results in our study [
1,
2,
3,
4,
8,
11,
12,
13,
16]. Patients resistant to initial therapy were mostly adolescents who presented with longer disease duration, severe thrombocytopenia, and severe bleeding manifestations, many of whom presented with comorbidities or autoantibodies and a positive family medical history (
Figure 2). Higher resistance levels were measured when corticosteroid therapy was administered rather than intravenous immunoglobulin therapy. All the aforementioned parameters can be found in the data from the literature as markers of chITP [
1,
3,
4].
Around 20% of patients tested positive for
Helicobacter pylori antigen detected in stool sample. To date, no mechanisms have been found that explain how
Helicobacter pylori affects ITP pathogenesis. Recent analyses show a correlation between
Helicobacter pylori infection and poor response to therapy in almost 50% of patients with ITP. Therefore, theory shows that eradication of the infection could contribute to the treatment of ndITP. On the other hand, eradication therapy for
Helicobacter pylori in chITP patients is less efficient for the treatment of thrombocytopenia. However, the results are still quite contradictory [
23]. In our study, the most common patients were young males who were asymptomatic or had mild bleeding manifestations, mild thrombocytopenia, and short duration of the disease. Unfortunately, the data on the number of patients who underwent esophagogastroduodenoscopy is absent. Medical history data show that eradication was completed in 21 of 33 patients, with no significant effect on the dominant disease (
Figure 3). There is no clear evidence that these patients would, nonetheless, spontaneously recover or that eradication therapy and elimination of the cause of disease would contribute to recovery. Exact connections between the two entities are still unknown [
23].
One-third of all platelets are stored in spleen sinusoids and function as reserve platelets. Old platelets are most commonly destroyed in the red pulp of the spleen. In the spleen, the pathophysiological mechanism of forming anti-thrombocyte antibodies occurs, which matches the location of platelet destruction in ITP patients. Thus, this organ deserves the greatest attention [
8,
11,
12,
24]. Using abdominal ultrasound, we detected splenomegaly in almost 25% of patients, with no significant effect on the outcome of disease. An occurrence of organomegaly excludes the diagnosis of ITP; however, this rule does not apply to chITP. The long-term destruction of platelets in the reticuloendothelial spleen system leads to their hyperplasia and occasionally causes organ enlargement depending on the activity of the immunological process. Other researchers confirm finding almost equal percentages of splenomegaly in their studies, especially those that opt for splenectomy as a therapy option for ITP [
8,
11,
12,
19,
24]. Furthermore, it was noted that around 14% of the patients had accessory spleen, which had no significant effect on the outcome of disease.
Upon the diagnosis of chITP, the mean platelet count was around 35 × 10
9/L, and the mean MPV was around 10.5 fL. All children with chITP underwent cytological examination of bone marrow aspirate, and no dysplastic changes were noted. Around 50% of patients had platelet counts between 20 and 50 × 10
9/L, and 40% of patients had <20 × 10
9/L (
Table 4). A higher platelet count of 30–50 × 10
9/L (depending on the author) at diagnosis of ITP is one of the important criteria for the onset of chITP [
3,
4]. On the other hand, in the course of chronic disease, around 85–90% of patients affected for more than 2 years presented with severe bleeding episodes (BSS 3 and 4), received many transfusions of blood derivatives, and had platelet counts of <20 × 10
9/L (
Table 5). Therefore, platelet count is the factor that determines the severity of bleeding manifestations in patients with chITP, as the results of other studies show [
3,
4,
8,
11,
12,
19,
25].
An interesting phenomenon is noted regarding MPV. All patients with extremely large platelets (MPV > 12 fL) and platelet counts of <20 × 10
9/L belong to the group of patients with mild bleeding manifestations (BSS 1 and 2) and increased production of megakaryocytes in the bone marrow. On the other hand, all patients with normal MPV (7–10 fL) and platelet counts of <20 × 10
9/L have severe bleedings (BSS 3 and 4) and rare megakaryocytes in the bone marrow. Therefore, the presence of larger, young, reactive platelets may compensate for the reduced platelet count, as they are more metabolically and hemostatically active [
26]. The relationship between MPV and bleeding is a significant topic for discussion. This can be attributed to the fact that the bone marrow gradually adapts and becomes chronically stimulated to produce younger, more reactive platelets on the one hand. On the other hand, the question arises regarding the influence and effect of autoantibodies on megakaryocytes. It is likely that in patients whose autoantibodies act solely on peripheral platelets, but not on megakaryocytes, the bone marrow remains capable of producing platelets efficiently. Conversely, in patients whose megakaryocytes are affected by autoantibodies, the production of smaller platelets occurs—platelets that are less active in hemostatic processes and, consequently, more prone to bleeding [
1,
20,
21,
22]. In such patients, bone marrow biopsies often reveal a reduced number of megakaryocytes, and these patients frequently present with ITP in combination with other autoimmune diseases [
2,
8,
11].
Interestingly, normal or increased production of megakaryocytes in the bone marrow shows no correlation with other examined parameters. On the other hand, all patients with rare megakaryocytes in the bone marrow have some accompanying autoimmune disease or test positive for autoantibodies. Most of the patients have severe bleeding manifestations and show resistance to initial therapy. Therefore, autoantibodies, apart from their influence on platelets, can bind to megakaryocytes, inhibit their maturation, or cause their destruction. Moreover, autoantibodies can affect cytokines that are necessary for the growth and proliferation of megakaryocytes [
1,
2,
8,
11,
27].
Almost 70% of patients had higher levels of LDH. It was noted that levels of LDH higher than 700 U/L correlated with a low platelet count (<20 × 10
9/L) and resistance to initial treatment. Since LDH is an intracellular enzyme that is released by increased platelet destruction, these levels were expected. Frequent viral infections during a patient’s childhood cannot represent an adequate parameter for the assessment of disease activity since they can usually increase LDH levels, as opposed to infections in adulthood [
5].
Only three patients with chITP had sufficient serum vitamin D levels, ranging from 30 to 40 ng/mL, whereas the remaining patients had lower serum vitamin D levels. The relation between these two entities has been an interesting topic in recent years. In some studies, the correlation was confirmed in more than 80%, which is similar to the results from our study. However, the evidence shows that vitamin D receptor gene polymorphism may be attributed to the disease rather than low levels of vitamin D alone. However, more extensive research should be conducted regarding this area [
28]. Nevertheless, vitamin D supplementation proves to be efficient in patients with ITP.
For approximately 45% of patients, only initial therapy was required in the first 12 months; afterwards, it was confirmed (according to the data from their medical histories) that the patients entered a stable phase. An additional 25% of patients also entered a stable phase after initial therapy and available second-line therapy were administered. These patients were either without or with mild signs of hemorrhagic syndrome; however, the treatment was not required until the second year in the duration of disease. Around 20% of patients underwent splenectomy as the remaining treatment method, and all of these patients did not have any bleeding episodes after this intervention. Unfortunately, 10% of children with chITP received all available treatments and splenectomy as part of conducted therapy and periodically still have bleeding episodes (
Figure 4). Most of the children from the last group are very severe cases, and the disease remains active after 5 years of treatment. This percentage is approximately similar to that cited in another study that dealt with this topic [
3,
4,
8,
11,
12,
19,
26].


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}