
Role of Next-Generation Sequencing in Diagnosis of Familial Hypercholesterolemia in Serbia

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population Sample
2.2. Laboratory Analyses
2.3. FH Diagnosis
2.4. Genetic Analysis
2.5. Statistical Analysis
3. Results
3.1. Study Population
3.2. Genetic Testing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASCVD | Atherosclerotic cardiovascular disease |
| APOB | Apolipoprotein B |
| CAD | Coronary artery disease |
| DFH | Definitive familial hypercholesterolemia |
| DLCN | Dutch Lipid Clinic Network Diagnostic Criteria |
| FH | Familial hypercholesterolemia |
| HGMD | Human Gene Mutation Database |
| HeFH | Heterozygous familial hypercholesterolemia |
| HoFH | Homozygous form of familial hypercholesterolemia |
| JAS | Japan Atherosclerosis Society |
| LDL | Low-density lipoprotein |
| LDLR | Low-density lipoprotein receptor |
| LDLRAP1 | Low-density lipoprotein receptor adapter protein 1 |
| NGS | Next-generation sequencing |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PFH | Probable familial hypercholesterolemia |
| VLDL | Very-low-density lipoprotein |
References
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Shah, N.P.; Ahmed, H.M.; Wilson Tang, W.H. Familial Hypercholesterolemia: Detect, Treat, and Ask about Family. Clevel. Clin. J. Med. 2020, 87, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Lui, D.T.W.; Lee, A.C.H.; Tan, K.C.B. Management of Familial Hypercholesterolemia: Current Status and Future Perspectives. J. Endocr. Soc. 2021, 5, bvaa122. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients with Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial Hypercholesterolaemia Is Underdiagnosed and Undertreated in the General Population: Guidance for Clinicians to Prevent Coronary Heart Disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef]
- Defesche, J.C.; Lansberg, P.J.; Umans-Eckenhausen, M.A.W.; Kastelein, J.J.P. Advanced Method for the Identification of Patients with Inherited Hypercholesterolemia. Semin. Vasc. Med. 2004, 4, 59–65. [Google Scholar] [CrossRef]
- Williams, R.R.; Hunt, S.C.; Schumacher, M.C.; Hegele, R.A.; Leppert, M.F.; Ludwig, E.H.; Hopkins, P.N. Diagnosing Heterozygous Familial Hypercholesterolemia Using New Practical Criteria Validated by Molecular Genetics. Am. J. Cardiol. 1993, 72, 171–176. [Google Scholar] [CrossRef]
- Risk of Fatal Coronary Heart Disease in Familial Hypercholesterolaemia. Scientific Steering Committee on Behalf of the Simon Broome Register Group. BMJ 1991, 303, 893–896. [CrossRef]
- Mikhailova, S.; Ivanoshchuk, D.; Timoshchenko, O.; Shakhtshneider, E. Genes Potentially Associated with Familial Hypercholesterolemia. Biomolecules 2019, 9, 807. [Google Scholar] [CrossRef]
- Cuchel, M.; Bruckert, E.; Ginsberg, H.N.; Raal, F.J.; Santos, R.D.; Hegele, R.A.; Kuivenhoven, J.A.; Nordestgaard, B.G.; Descamps, O.S.; Steinhagen-Thiessen, E.; et al. Homozygous Familial Hypercholesterolaemia: New Insights and Guidance for Clinicians to Improve Detection and Clinical Management. A Position Paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014, 35, 2146–2157. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a Comprehensive Mutation Repository for Clinical and Molecular Genetics, Diagnostic Testing and Personalized Genomic Medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef]
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: A HuGE Prevalence Review. Am. J. Epidemiol. 2004, 160, 407–420. [Google Scholar] [CrossRef]
- Fernández-Higuero, J.A.; Etxebarria, A.; Benito-Vicente, A.; Alves, A.C.; Arrondo, J.L.R.; Ostolaza, H.; Bourbon, M.; Martin, C. Structural Analysis of APOB Variants, p.(Arg3527Gln), p.(Arg1164Thr) and p.(Gln4494del), Causing Familial Hypercholesterolaemia Provides Novel Insights into Variant Pathogenicity. Sci. Rep. 2015, 5, 18184. [Google Scholar] [CrossRef]
- Guo, Q.; Feng, X.; Zhou, Y. PCSK9 Variants in Familial Hypercholesterolemia: A Comprehensive Synopsis. Front. Genet. 2020, 11, 1020. [Google Scholar] [CrossRef]
- Pham, N.H.; Truong, P.K.; Lao, T.D.; Le, T.A.H. Proprotein Convertase Subtilisin/Kexin Type 9 Gene Variants in Familial Hypercholesterolemia: A Systematic Review and Meta-Analysis. Processes 2021, 9, 283. [Google Scholar] [CrossRef]
- Shaik, N.A.; Al-Qahtani, F.; Nasser, K.; Jamil, K.; Alrayes, N.M.; Elango, R.; Awan, Z.A.; Banaganapalli, B. Molecular Insights into the Coding Region Mutations of Low-Density Lipoprotein Receptor Adaptor Protein 1 (LDLRAP1) Linked to Familial Hypercholesterolemia. J. Gene Med. 2020, 22, e3176. [Google Scholar] [CrossRef]
- Leigh, T.; Kawai, T.; Preston, K.; Kelemen, S.; Okune, R.; St Paul, A.; Corbett, C.; Peluzzo, A.M.; Yu, J.; Scalia, R.G.; et al. Deletion of LDLRAP1 Induces Atherosclerotic Plaque Formation, Insulin Resistance, and Dysregulated Insulin Response in Adipose Tissue. Am. J. Pathol. 2022, 192, 1092–1108. [Google Scholar] [CrossRef]
- Watts, G.F.; Gidding, S.; Wierzbicki, A.S.; Toth, P.P.; Alonso, R.; Brown, W.V.; Bruckert, E.; Defesche, J.; Lin, K.K.; Livingston, M.; et al. Integrated Guidance on the Care of Familial Hypercholesterolaemia from the International FH Foundation. Int. J. Cardiol. 2014, 171, 309–325. [Google Scholar] [CrossRef]
- Kumar, V.; Gill, K.D. Estimation of Blood Glucose Levels by Glucose Oxidase Method. In Basic Concepts in Clinical Biochemistry: A Practical Guide; Kumar, V., Gill, K.D., Eds.; Springer: Singapore, 2018; pp. 57–60. ISBN 978-981-10-8186-6. [Google Scholar]
- Versmissen, J.; Oosterveer, D.M.; Yazdanpanah, M.; Defesche, J.C.; Basart, D.C.G.; Liem, A.H.; Heeringa, J.; Witteman, J.C.; Lansberg, P.J.; Kastelein, J.J.P.; et al. Efficacy of Statins in Familial Hypercholesterolaemia: A Long Term Cohort Study. BMJ 2008, 337, a2423. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Tsimikas, S.; Fazio, S. The Severe Hypercholesterolemia Phenotype: Clinical Diagnosis, Management, and Emerging Therapies. J. Am. Coll. Cardiol. 2014, 63, 1935–1947. [Google Scholar] [CrossRef]
- Berberich, A.J.; Hegele, R.A. The Complex Molecular Genetics of Familial Hypercholesterolaemia. Nat. Rev. Cardiol. 2019, 16, 9–20. [Google Scholar] [CrossRef]
- Etxebarria, A.; Palacios, L.; Stef, M.; Tejedor, D.; Uribe, K.B.; Oleaga, A.; Irigoyen, L.; Torres, B.; Ostolaza, H.; Martin, C. Functional Characterization of Splicing and Ligand-Binding Domain Variants in the LDL Receptor. Hum. Mutat. 2012, 33, 232–243. [Google Scholar] [CrossRef]
- Chora, J.R.; Iacocca, M.A.; Tichý, L.; Wand, H.; Kurtz, C.L.; Zimmermann, H.; Leon, A.; Williams, M.; Humphries, S.E.; Hooper, A.J.; et al. The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel Consensus Guidelines for LDLR Variant Classification. Genet. Med. 2022, 24, 293–306. [Google Scholar] [CrossRef]
- Bertolini, S.; Pisciotta, L.; Rabacchi, C.; Cefalù, A.B.; Noto, D.; Fasano, T.; Signori, A.; Fresa, R.; Averna, M.; Calandra, S. Spectrum of Mutations and Phenotypic Expression in Patients with Autosomal Dominant Hypercholesterolemia Identified in Italy. Atherosclerosis 2013, 227, 342–348. [Google Scholar] [CrossRef]
- de Paiva Silvino, J.P.; Jannes, C.E.; Tada, M.T.; Lima, I.R.; Silva, I.d.F.O.; Pereira, A.C.; Gomes, K.B. Cascade Screening and Genetic Diagnosis of Familial Hypercholesterolemia in Clusters of the Southeastern Region from Brazil. Mol. Biol. Rep. 2020, 47, 9279–9288. [Google Scholar] [CrossRef]
- Madar, L.; Juhász, L.; Szűcs, Z.; Kerkovits, L.; Harangi, M.; Balogh, I. Establishing the Mutational Spectrum of Hungarian Patients with Familial Hypercholesterolemia. Genes 2022, 13, 153. [Google Scholar] [CrossRef]
- Mollaki, V.; Drogari, E. Genetic Causes of Monogenic Familial Hypercholesterolemia in the Greek Population: Lessons, Mistakes, and the Way Forward. J. Clin. Lipidol. 2016, 10, 748–756. [Google Scholar] [CrossRef]
- Lombardi, M.P.; Redeker, E.J.; Defesche, J.C.; Kamerling, S.W.; Trip, M.D.; Mannens, M.M.; Havekes, L.M.; Kastelein, J.J. Molecular Genetic Testing for Familial Hypercholesterolemia: Spectrum of LDL Receptor Gene Mutations in The Netherlands. Clin. Genet. 2000, 57, 116–124. [Google Scholar] [CrossRef]
- Mozas, P.; Castillo, S.; Tejedor, D.; Reyes, G.; Alonso, R.; Franco, M.; Saenz, P.; Fuentes, F.; Almagro, F.; Mata, P.; et al. Molecular Characterization of Familial Hypercholesterolemia in Spain: Identification of 39 Novel and 77 Recurrent Mutations in LDLR. Hum. Mutat. 2004, 24, 187. [Google Scholar] [CrossRef]
- Turkyilmaz, A.; Kurnaz, E.; Alavanda, C.; Yarali, O.; Kartal Baykan, E.; Yavuz, D.; Cayir, A.; Ata, P. The Spectrum of Low-Density Lipoprotein Receptor Mutations in a Large Turkish Cohort of Patients with Familial Hypercholesterolemia. Metab. Syndr. Relat. Disord. 2021, 19, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Larrea-Sebal, A.; Alonso-Estrada, R.; Aguilo-Arce, J.; Ostolaza, H.; Palacios, L.; Martin, C. Mutation Type Classification and Pathogenicity Assignment of Sixteen Missense Variants Located in the EGF-Precursor Homology Domain of the LDLR. Sci. Rep. 2020, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Phuong Kim, T.; Thuan Duc, L.; Thuy Ai, H.L. The Major Molecular Causes of Familial Hypercholesterolemia. Asian J. Pharm. Res. Health Care 2018, 10, 60–68. [Google Scholar] [CrossRef]
- Rieck, L.; Bardey, F.; Grenkowitz, T.; Bertram, L.; Helmuth, J.; Mischung, C.; Spranger, J.; Steinhagen-Thiessen, E.; Bobbert, T.; Kassner, U.; et al. Mutation Spectrum and Polygenic Score in German Patients with Familial Hypercholesterolemia. Clin. Genet. 2020, 98, 457–467. [Google Scholar] [CrossRef]
- Mollaki, V.; Progias, P.; Drogari, E. Familial Hypercholesterolemia in Greek Children and Their Families: Genotype-to-Phenotype Correlations and a Reconsideration of LDLR Mutation Spectrum. Atherosclerosis 2014, 237, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Sjouke, B.; Hovingh, G.K.; Kastelein, J.J.P.; Stefanutti, C. Homozygous Autosomal Dominant Hypercholesterolaemia: Prevalence, Diagnosis, and Current and Future Treatment Perspectives. Curr. Opin. Lipidol. 2015, 26, 200–209. [Google Scholar] [CrossRef]
- Hu, M.; Hooper, A.J.; van Bockxmeer, F.M.; Watts, G.F.; Chan, J.C.; Tomlinson, B. Management of Familial Hypercholesterolemia in Hong Kong. J. Atheroscler. Thromb. 2016, 23, 520–531. [Google Scholar] [CrossRef]
- Raal, F.J.; Honarpour, N.; Blom, D.J.; Hovingh, G.K.; Xu, F.; Scott, R.; Wasserman, S.M.; Stein, E.A. TESLA Investigators Inhibition of PCSK9 with Evolocumab in Homozygous Familial Hypercholesterolaemia (TESLA Part B): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2015, 385, 341–350. [Google Scholar] [CrossRef]
- Marusic, T.; Sustar, U.; Sadiq, F.; Kotori, V.; Mlinaric, M.; Kovac, J.; Shafi, S.; Khan, I.; Cevc, M.; Trebusak Podkrajsek, K.; et al. Genetic and Clinical Characteristics of Patients with Homozygous and Compound Heterozygous Familial Hypercholesterolemia From Three Different Populations: Case Series. Front. Genet. 2020, 11, 572176. [Google Scholar] [CrossRef]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular Genetics of the LDL Receptor Gene in Familial Hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Michaely, P. The Epidermal Growth Factor Homology Domain of the LDL Receptor Drives Lipoprotein Release through an Allosteric Mechanism Involving H190, H562, and H586. J. Biol. Chem. 2008, 283, 26528–26537. [Google Scholar] [CrossRef] [PubMed]
- Tybjaerg-Hansen, A.; Humphries, S.E. Familial Defective Apolipoprotein B-100: A Single Mutation That Causes Hypercholesterolemia and Premature Coronary Artery Disease. Atherosclerosis 1992, 96, 91–107. [Google Scholar] [CrossRef]
- Rodríguez-Jiménez, C.; de la Peña, G.; Sanguino, J.; Poyatos-Peláez, S.; Carazo, A.; Martínez-Hernández, P.L.; Arrieta, F.; Mostaza, J.M.; Gómez-Coronado, D.; Rodríguez-Nóvoa, S. Identification and Functional Analysis of APOB Variants in a Cohort of Hypercholesterolemic Patients. Int. J. Mol. Sci. 2023, 24, 7635. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Lee, I.; Zhu, W.; Arnold, K.; Taylor, S.; Innerarity, T.L. Identification of the Low Density Lipoprotein Receptor-Binding Site in Apolipoprotein B100 and the Modulation of Its Binding Activity by the Carboxyl Terminus in Familial Defective Apo-B100. J. Clin. Investig. 1998, 101, 1084–1093. [Google Scholar] [CrossRef]
- Brænne, I.; Kleinecke, M.; Reiz, B.; Graf, E.; Strom, T.; Wieland, T.; Fischer, M.; Kessler, T.; Hengstenberg, C.; Meitinger, T.; et al. Systematic Analysis of Variants Related to Familial Hypercholesterolemia in Families with Premature Myocardial Infarction. Eur. J. Hum. Genet. 2016, 24, 191–197. [Google Scholar] [CrossRef]
- Truong, P.K.; Van Bui, C.; Lao, T.D.; Le, T.H.A. Detection of Defective Apolipoprotein B-100 R3500Q Mutation Caused Familial Hypercholesterolemia in Vietnamese Patients. In Proceedings of the 6th International Conference on the Development of Biomedical Engineering in Vietnam (BME6); Vo Van, T., Nguyen Le, T.A., Nguyen Duc, T., Eds.; Springer: Singapore, 2018; pp. 275–279. [Google Scholar]
- Futema, M.; Whittall, R.A.; Kiley, A.; Steel, L.K.; Cooper, J.A.; Badmus, E.; Leigh, S.E.; Karpe, F.; Neil, H.A.W.; Humphries, S.E. Analysis of the Frequency and Spectrum of Mutations Recognised to Cause Familial Hypercholesterolaemia in Routine Clinical Practice in a UK Specialist Hospital Lipid Clinic. Atherosclerosis 2013, 229, 161–168. [Google Scholar] [CrossRef]
- Abifadel, M.; Boileau, C. Genetic and Molecular Architecture of Familial Hypercholesterolemia. J. Intern. Med. 2023, 293, 144–165. [Google Scholar] [CrossRef]
- Martín-Campos, J.M. Genetic Determinants of Plasma Low-Density Lipoprotein Cholesterol Levels: Monogenicity, Polygenicity, and “Missing” Heritability. Biomedicines 2021, 9, 1728. [Google Scholar] [CrossRef]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; Whittall, R.A.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of Variant Selection for the LDL Cholesterol Genetic Risk Score in the Diagnosis of the Polygenic Form of Clinical Familial Hypercholesterolemia and Replication in Samples from 6 Countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef]
- Amor-Salamanca, A.; Castillo, S.; Gonzalez-Vioque, E.; Dominguez, F.; Quintana, L.; Lluís-Ganella, C.; Escudier, J.M.; Ortega, J.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Genetically Confirmed Familial Hypercholesterolemia in Patients with Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2017, 70, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Okada, H.; Nomura, A.; Usui, S.; Sakata, K.; Nohara, A.; Yamagishi, M.; Takamura, M.; Kawashiri, M.-A. Clinical Diagnostic Criteria of Familial Hypercholesterolemia—A Comparison of the Japan Atherosclerosis Society and Dutch Lipid Clinic Network Criteria. Circ. J. 2021, 85, 891–897. [Google Scholar] [CrossRef] [PubMed]
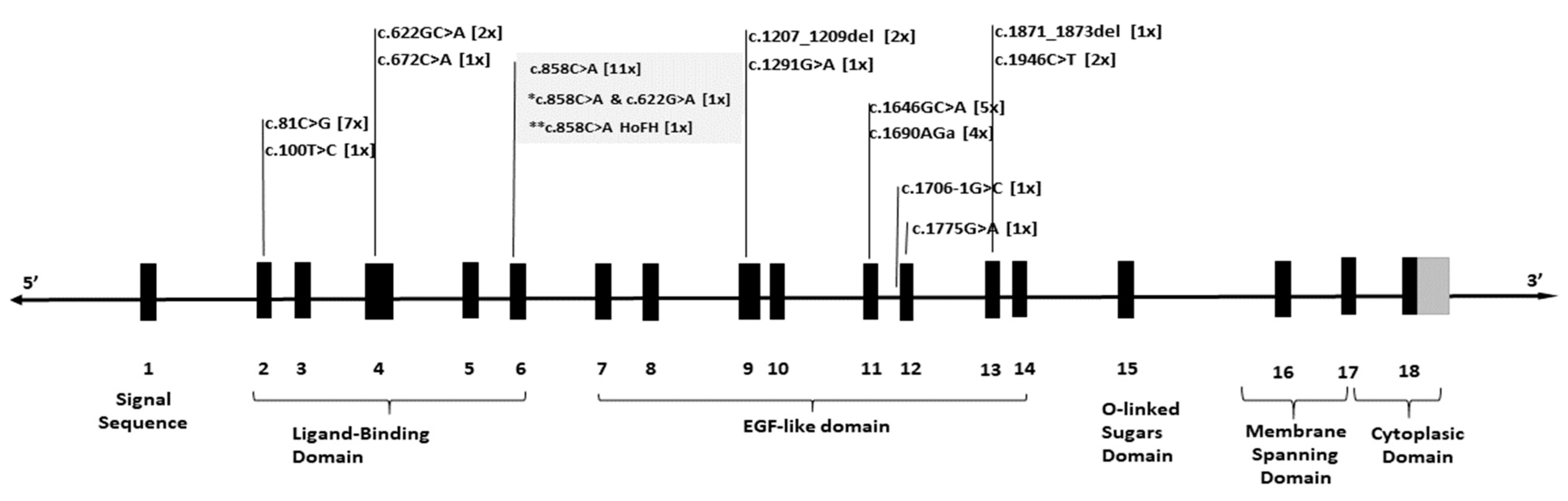

{kind=link}
| Diagnosis (DLCN Criteria) | No. of FH Patients (DLCN Classification) | No. of Genetically Confirmed | % of Genetically Confirmed |
|---|---|---|---|
| Definite | 31 | 26 | 83.9% |
| Probable | 10 | 2 | 20.0% |
| Possible | 32 | 9 | 28.1% |
| Unlikely | 28 | 7 | 25.0% |
| Clinical Parameters Being Measured (Mean ± SD) | All Patients (n = 101) | Genetically FH-Negative Patients (n = 57) | Genetically FH-Positive Patients (n = 44) | LDLR-Positive Patients (n = 41) | LDLR-Positive Patients Without the c.858C>A p.(Ser286Arg) Variant (n = 28) | LDLR-Positive Patients with the c.858C>A p.(Ser286Arg) (n = 13) | LDLR-Positive Patients with the c.858C>A p.(Ser286Arg) Variant Excluding HoFH/CoFH (n = 11) | APOB-Positive Patients (n = 3) |
|---|---|---|---|---|---|---|---|---|
| Age (years) | 55.1 ± 15.8 | 60.8 ± 14.9 * | 48.0 ±14.1 * | 48.5 ± 14.3 | 47.8 ±16.3 | 50.0 ± 9.26 | 49.1 ± 8.98 | 40.0 ± 10.6 |
| Gender (F/M) | 57/44 | 32/25 | 25/19 | 17/24 | 15/13 | 9/4 | 8/3 | 1/2 |
| TC (mmol/L) | 7.8 ± 2.8 | 6.7 ± 2.0 * | 9.3 ± 3.1 * | 9.3 ± 3.2 | 9.7 ± 3.5 | 8.5 ± 2.4 | 8.04 ± 1.94 | 8.30 ± 0.96 |
| LDL-C (mmol/L) | 5.4 ± 2.2 | 4.4 ± 1.7 * | 6.7 ± 2.3 * | 6.7 ± 2.3 | 6.9 ± 2.4 | 6.2 ± 2.2 | 5.69 ± 1.94 | 6.50 ± 1.1 |
| HDL-C (mmol/L) | 1.3 ± 0.34 | 1.3 ± 0.28 | 1.4 ± 0.4 | 1.4 ± 0.41 | 1.4 ± 0.43 | 1.4 ± 0.39 | 1.49 ± 0.38 | 1.2 ± 0.11 |
| TGs (mmol/L) | 1.9 ± 1.1 | 2.1 ± 0.98 * | 1.6 ± 1.2 * | 1.6 ± 1.2 | 1.7 ± 1.4 | 1.5 ± 0.75 | 1.57 ± 0.8 | 1.4 ± 0.21 |
| Glycemia (mmol/L) | 5.6 ± 1.7 | 5.7 ± 1.3 | 5.5 ± 2.1 | 5.6 ± 2.2 | 5.4 ± 2.0 | 5.9 ± 2.6 | 5.34 ± 0.76 | 4.90 ± 0.36 |
| BMI (kg/m2) | 25 ± 3.7 | 26 ± 3.1 | 25.0 ± 4.4 | 25.0 ± 4.5 | 24.0 ± 4.3 | 27 ± 4.90 | 26.2 ± 5.13 | 24.0 ± 1.20 |
| No. P. | Exon Number | Gene | Zygosity | Variant | Variant Type | ACMG Classification | Previous Description |
|---|---|---|---|---|---|---|---|
| 7 | 2 | LDLR | het | c.81C>G p.(Cys27Trp) | ms | P | ClinVar 226304 |
| 1 | 2 | LDLR | het | c.100T>C p.(Cys34Arg) | ms | P | ClinVar 251018, PMID: 33231818 |
| 2 | 4 | LDLR | het | c.622G>A p.(Glu208Lys) | ms | P | ClinVar 251328 |
| 1 | 4 | LDLR | het | c.672C>A p.(Asp224Glu) | ms | P | ClinVar 375790 |
| 11 | 6 | LDLR | het | c.858C>A p.(Ser286Arg) | ms | P | ClinVar 251488 |
| 1 | 6 | LDLR | hom | c.858C>A p.(Ser286Arg) | ms | P | ClinVar 251488 |
| 1 | 6 | LDLR | Comp. het | c.858C>A p.(Ser286Arg) | ms | P | ClinVar 251488 |
| 4 | c.622G>A p.(Glu208Lys) | ms | P | ClinVar 251328 | |||
| 2 | 9 | LDLR | het | c.1207_1209del p.(Phe403del) | ind | P | ClinVar 251731 |
| 1 | 9 | LDLR | het | c.1291G>A p.(Ala431Thr) | ms | P | ClinVar 3695 |
| 5 | 11 | LDLR | het | c.1646G>A p.(Gly549Asp) | ms | P | ClinVar 3698 |
| 4 | 11 | LDLR | het | c.1690A>G p.(Asn564Asp) | ms | P | ClinVar 251973 |
| 1 | intron 11 | LDLR | het | c.1706-1G>A | ss | P | ClinVar 251992 |
| 1 | 12 | LDLR | het | c.1775G>A p.(Gly592Glu) | ms | P | ClinVar 161271 |
| 1 | 13 | LDLR | het | c.1871_1873del p.(Ile624del) | ifd | P | ClinVar 250297 |
| 2 | 13 | LDLR | het | c.1946C>T p.(Pro649Leu) | ms | P | ClinVar 252122 |
| 3 | 26 | APOB | het | c.10580G>A p.(Arg3527Gln) | ms | P | ClinVar 17890 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukac, S.S.; Gasic, V.; Komazec, J.; Grubisa, I.; Popovic, L.; Rasulic, I.; Pavlovic, S.; Lalic, K. Role of Next-Generation Sequencing in Diagnosis of Familial Hypercholesterolemia in Serbia. Diagnostics 2025, 15, 1212. https://doi.org/10.3390/diagnostics15101212
Lukac SS, Gasic V, Komazec J, Grubisa I, Popovic L, Rasulic I, Pavlovic S, Lalic K. Role of Next-Generation Sequencing in Diagnosis of Familial Hypercholesterolemia in Serbia. Diagnostics. 2025; 15(10):1212. https://doi.org/10.3390/diagnostics15101212
Chicago/Turabian StyleLukac, Sandra Singh, Vladimir Gasic, Jovana Komazec, Ivana Grubisa, Ljiljana Popovic, Iva Rasulic, Sonja Pavlovic, and Katarina Lalic. 2025. "Role of Next-Generation Sequencing in Diagnosis of Familial Hypercholesterolemia in Serbia" Diagnostics 15, no. 10: 1212. https://doi.org/10.3390/diagnostics15101212
APA StyleLukac, S. S., Gasic, V., Komazec, J., Grubisa, I., Popovic, L., Rasulic, I., Pavlovic, S., & Lalic, K. (2025). Role of Next-Generation Sequencing in Diagnosis of Familial Hypercholesterolemia in Serbia. Diagnostics, 15(10), 1212. https://doi.org/10.3390/diagnostics15101212