
Novel Variants Linked to the Prodromal Stage of Parkinson’s Disease (PD) Patients

Abstract
1. Introduction
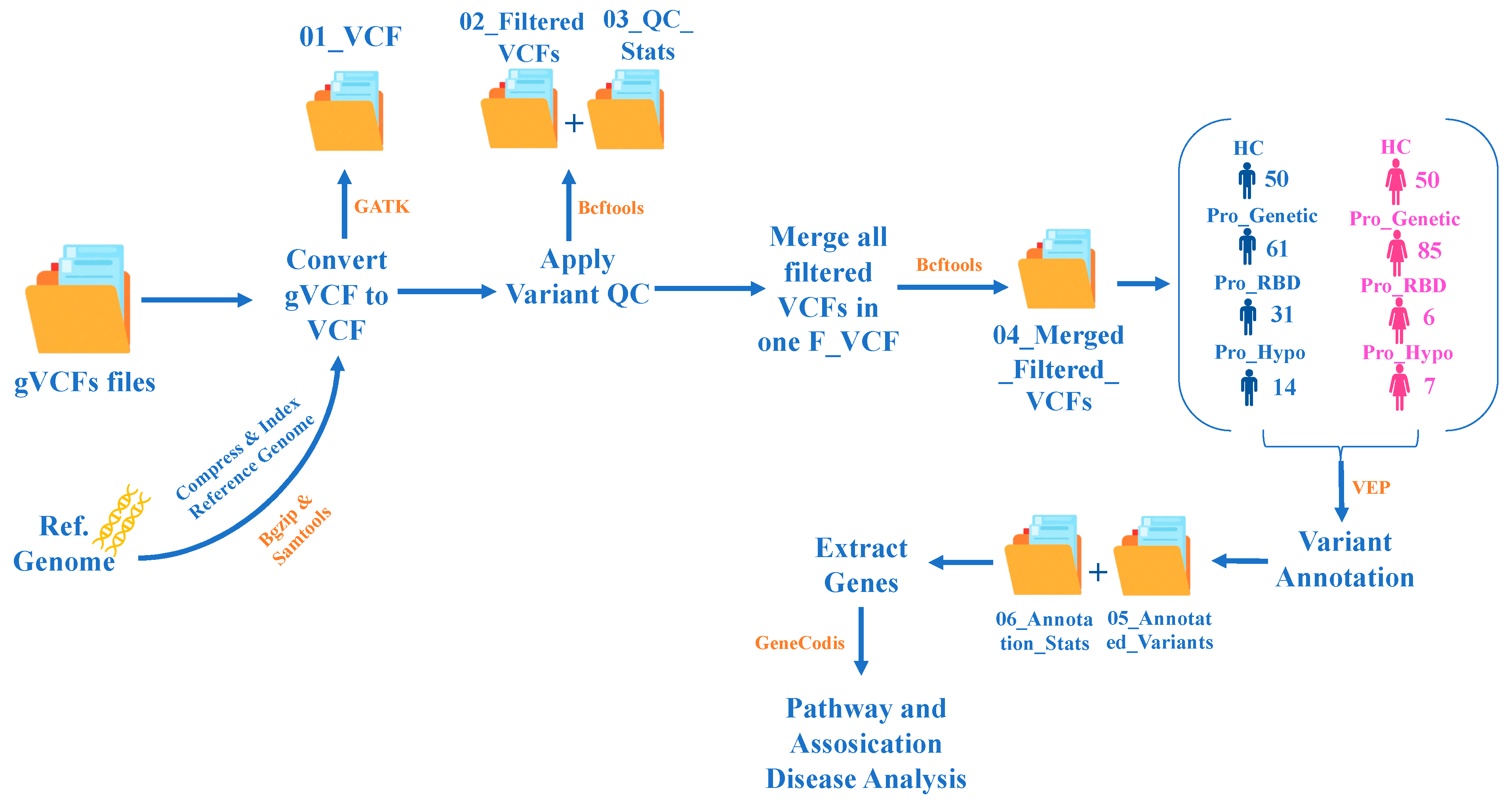
2. Materials and Methods
2.1. PPMI—Data Collection
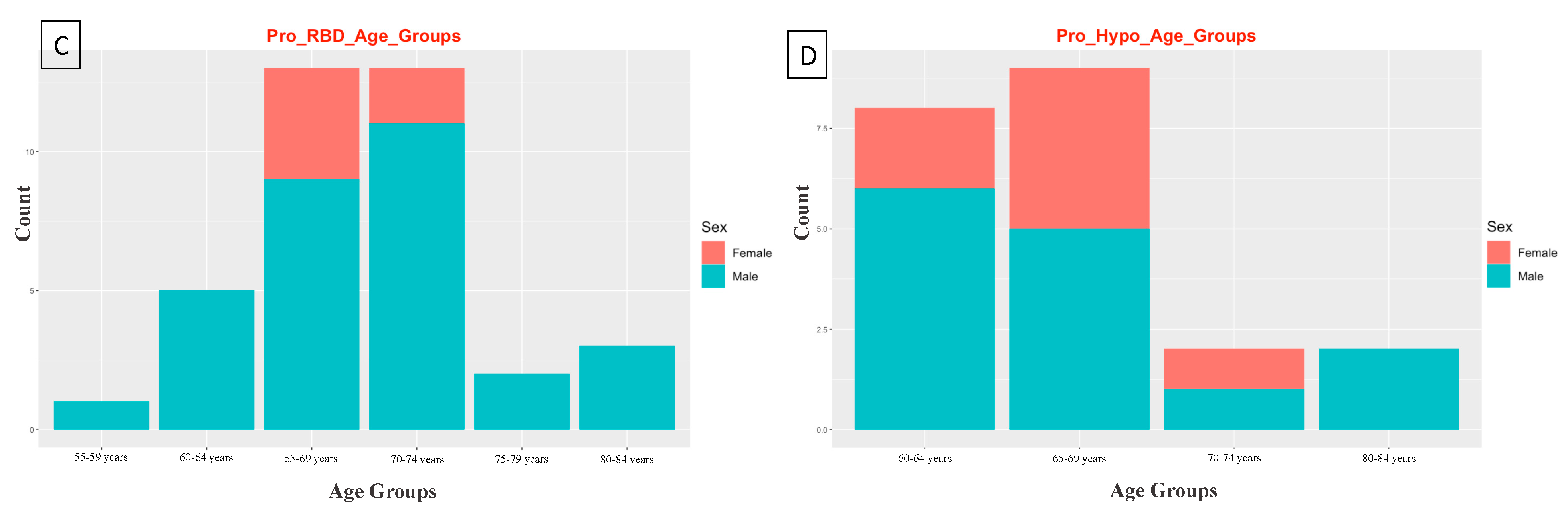
2.2. PPMI—Study Participants
2.3. Study Design
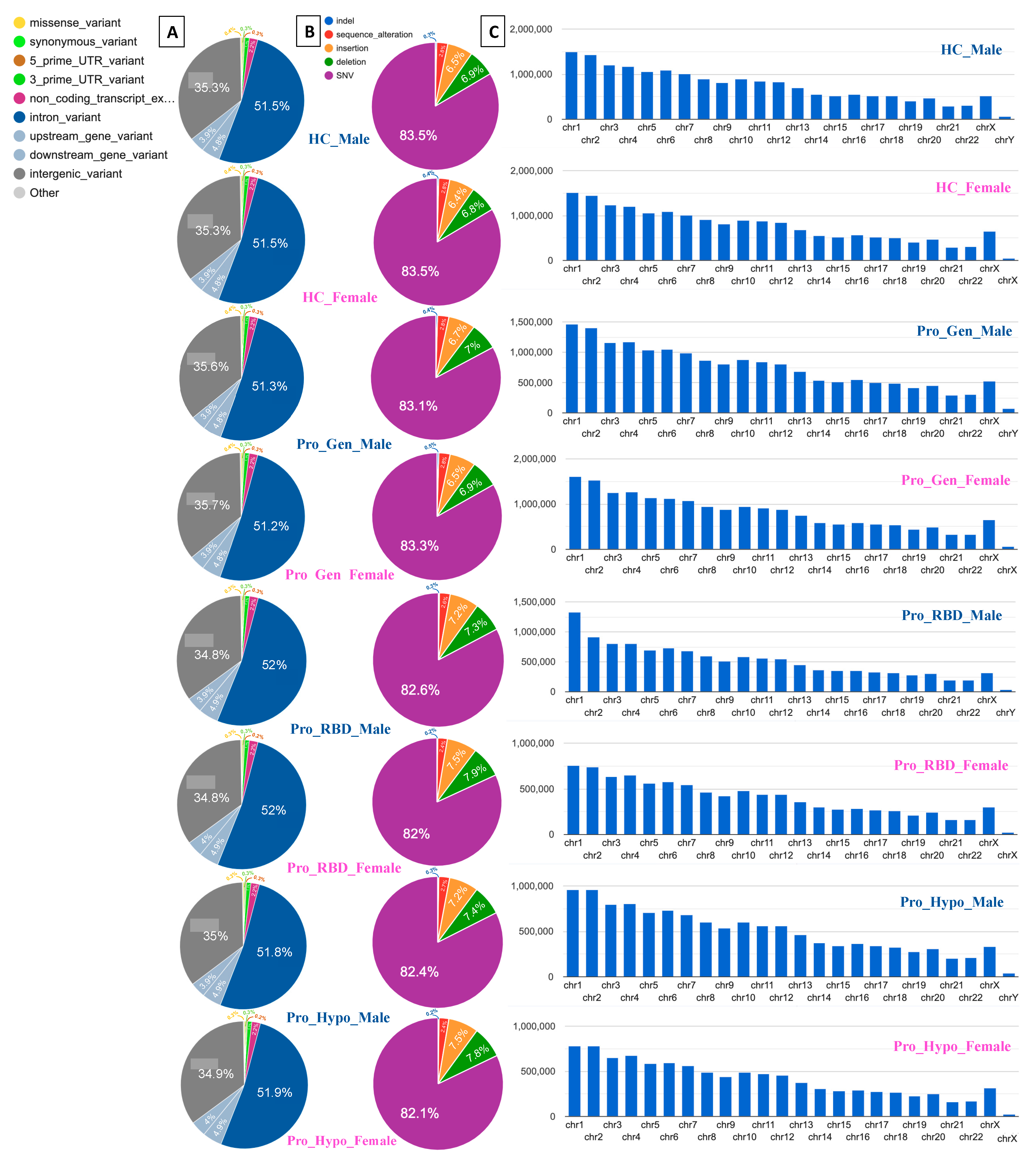
3. High Percentage of Intronic and Intergenic Variants
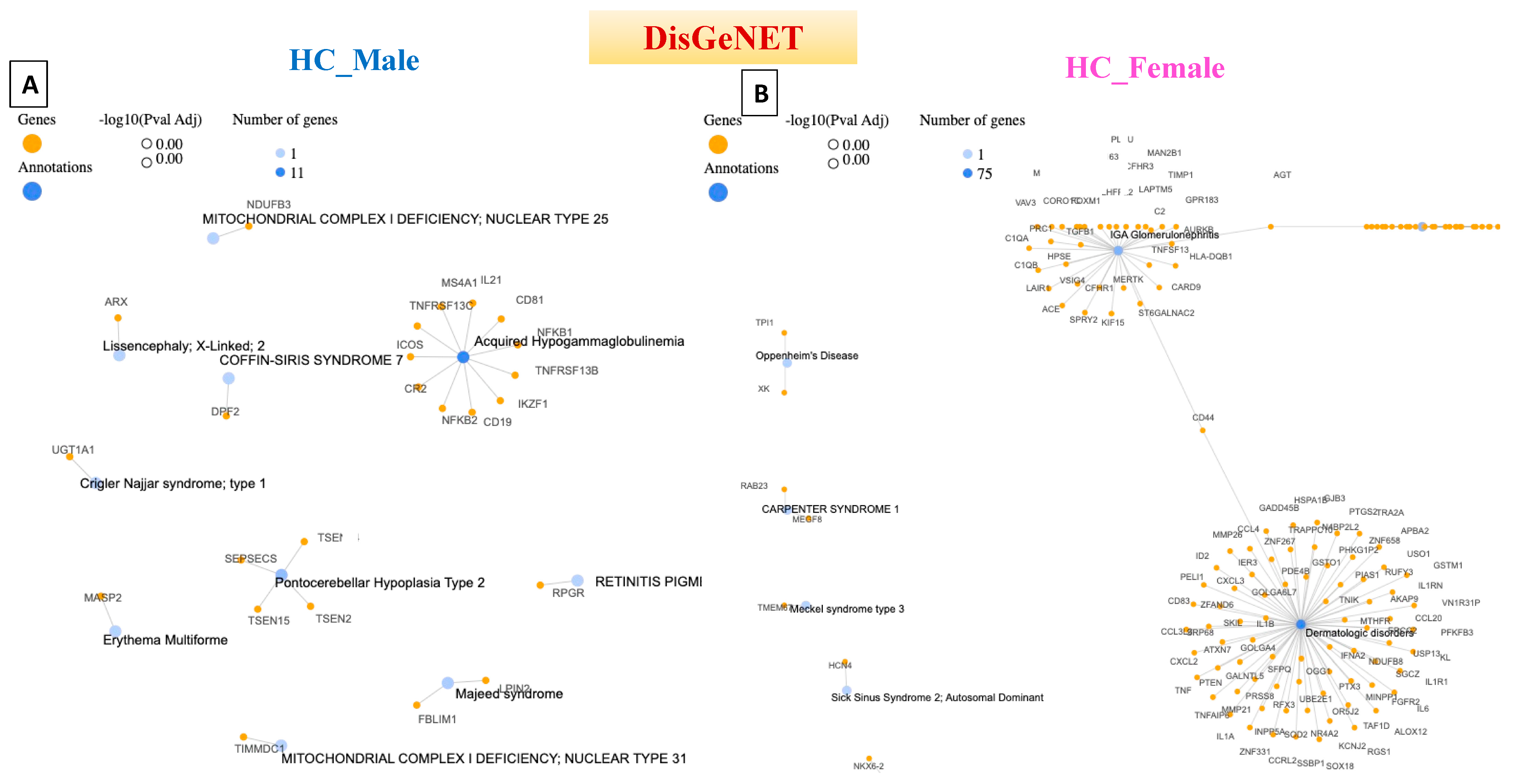
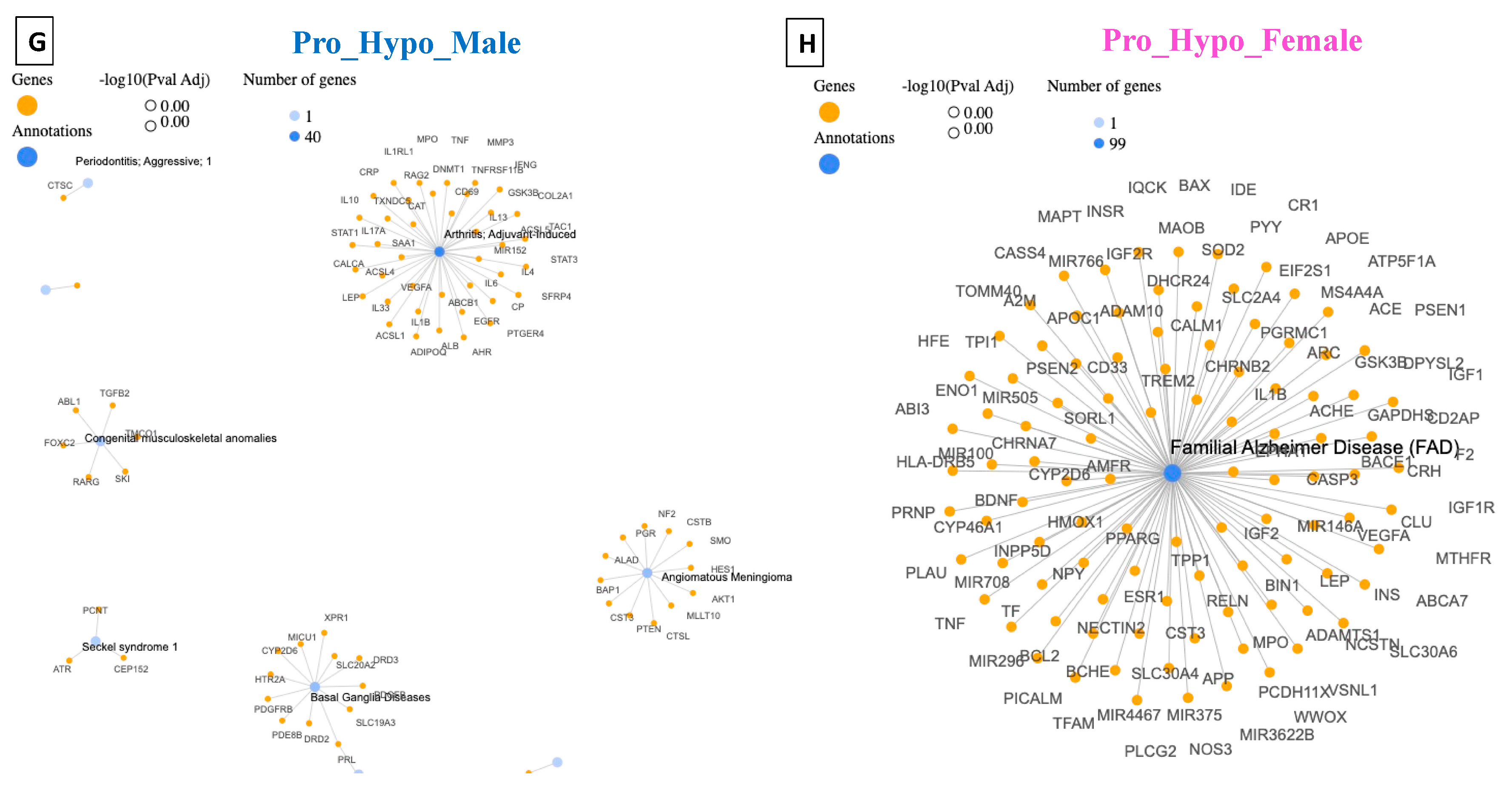
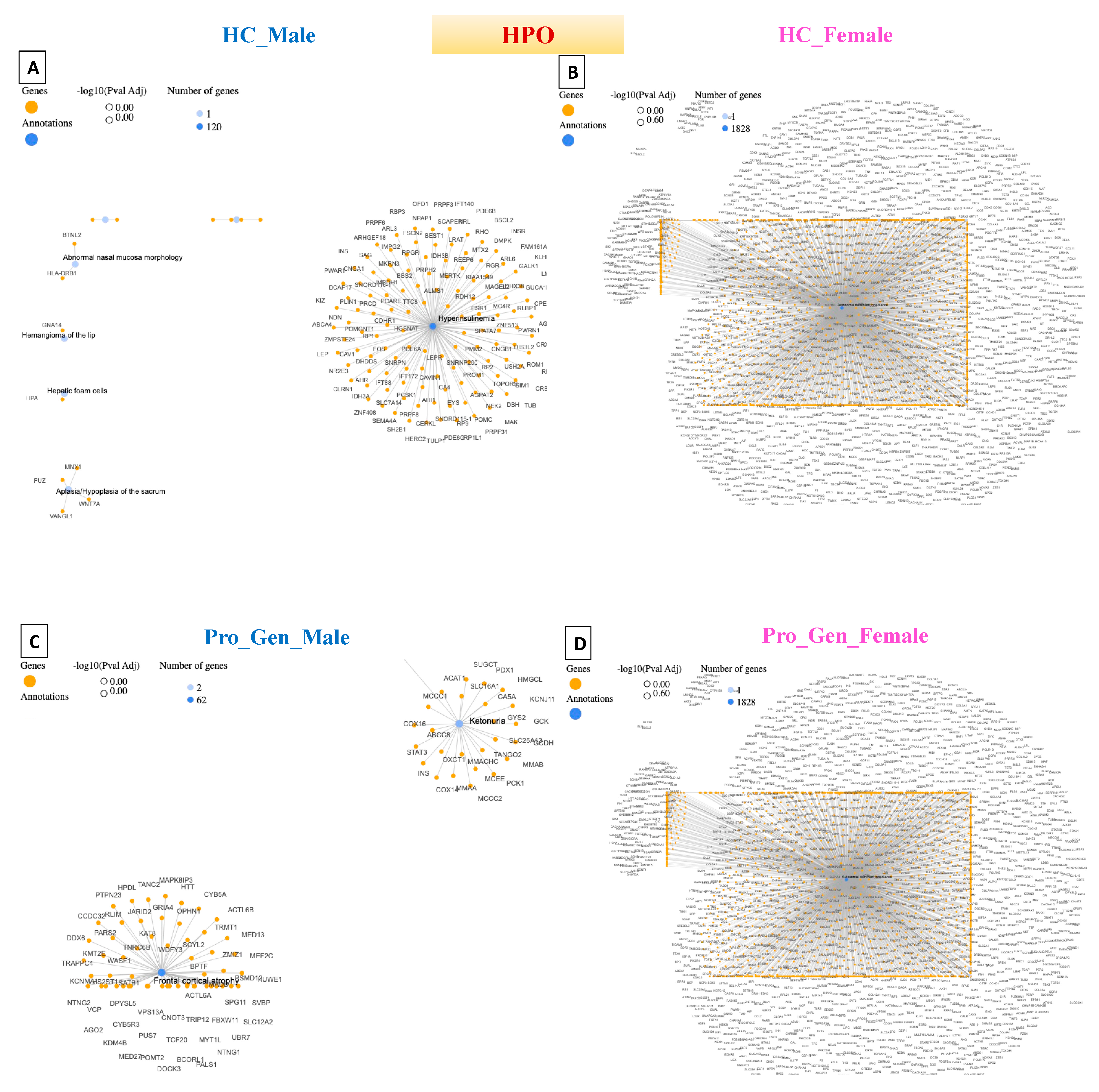
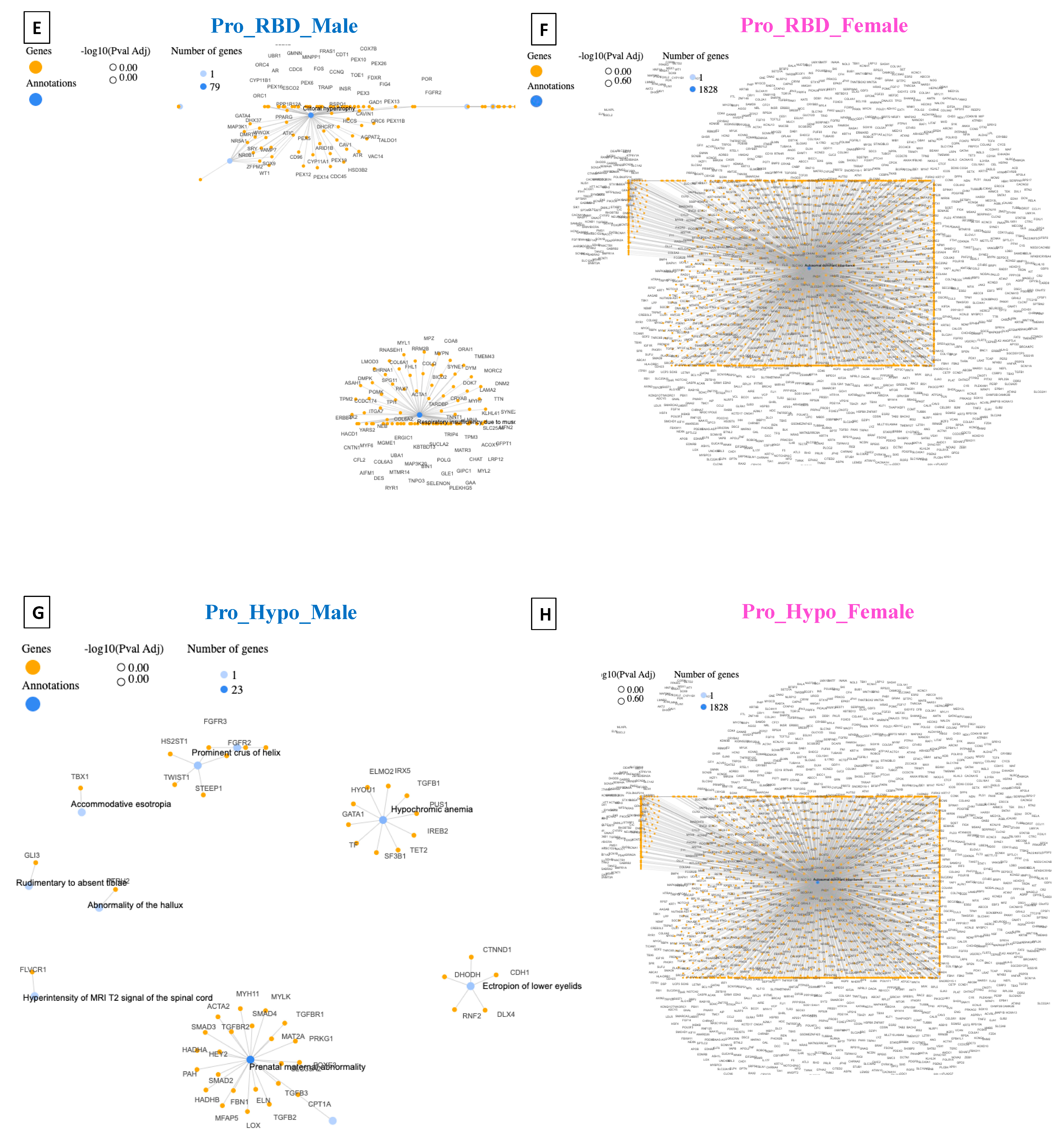
4. Disease–Gene Network (DisGeNET) Detection in Prodromal PD Populations
5. Detection of Human Phenotype Ontology (HPO) Data in Prodromal PD Populations
6. Online Mendelian Inheritance in Man (OMIM) Detection in Prodromal PD Populations
7. Novel Gene Detection in Prodromal PD Populations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.J.; Vitale, D.; Otani, D.V.; Lian, M.M.; Heilbron, K.; Iwaki, H.; Lake, J.; Solsberg, C.W.; Leonard, H.; Makarious, M.B. Multi-ancestry genome-wide association meta-analysis of Parkinson’s disease. Nat. Genet. 2023, 56, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Aarsland, D.; Barone, P.; Burn, D.J.; Hawkes, C.H.; Oertel, W.; Ziemssen, T. Identifying prodromal Parkinson’s disease: Pre-motor disorders in Parkinson’s disease. Mov. Disord. 2012, 27, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Schüle, B.; Rees, L.; Nichols, R.J.; Barlow, C. Multisystem Lewy body disease and the other parkinsonian disorders. Nat. Genet. 2015, 47, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Sci. Signal. 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Van Der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Divya, K.P.; Kandadai, R.M.; Yadav, R.; Satagopam, V.P.; Madhusoodanan, U.K.; Agarwal, P.; Kumar, N.; Ferreira, T.; Kumar, H.; et al. Genetic architecture of Parkinson’s disease in the Indian population: Harnessing genetic diversity to address critical gaps in Parkinson’s disease research. Front. Neurol. 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.; Jennings, D.; Lasch, S.; Siderowf, A.; Tanner, C.; Simuni, T.; Coffey, C.; Kieburtz, K.; Flagg, E.; Chowdhury, S.; et al. The Parkinson progression marker initiative (PPMI). Prog. Neurobiol. 2011, 95, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, Ș.; Falup-Pecurariu, O.; Țînț, D.; Falup-Pecurariu, C. REM sleep behaviour disorder in Parkinson’s disease. Exp. Ther. Med. 2021, 22, 812. [Google Scholar] [CrossRef] [PubMed]
- Tekriwal, A.; Kern, D.S.; Tsai, J.; Ince, N.F.; Wu, J.; Thompson, J.A.; Abosch, A. REM sleep behaviour disorder: Prodromal and mechanistic insights for Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 445–451. [Google Scholar] [CrossRef]
- Haehner, A.; Masala, C.; Walter, S.; Reichmann, H.; Hummel, T. Incidence of Parkinson’s disease in a large patient cohort with idiopathic smell and taste loss. J. Neurol. 2019, 266, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; McLean, C.Y.; Rick, J.; Eberly, S.; Hutten, S.J.; Gwinn, K.; Sutherland, M.; Martinez, M.; Heutink, P.; Williams, N.M.; et al. Diagnosis of Parkinson’s disease on the basis of clinical and genetic classification: A population-based modelling study. Lancet Neurol. 2015, 14, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Rigau, M.; Juan, D.; Valencia, A.; Rico, D. Intronic CNVs and gene expression variation in human populations. PLoS Genet. 2019, 15, e1007902. [Google Scholar] [CrossRef] [PubMed]
- Human-Genome-Structural-Variation-Working-Group. Completing the map of human genetic variation. Nature 2007, 447, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F. REM sleep behavior disorder: Updated review of the core features, the REM sleep behavior disorder-neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann. N. Y. Acad. Sci. 2010, 1184, 15–54. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Everaers, R. Structure and dynamics of interphase chromosomes. PLoS Comput. Biol. 2008, 4, e1000153. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, gkw943. [Google Scholar] [CrossRef]
- García-Moreno, A.; López-Domínguez, R.; Ramirez-Mena, A.; Pascual-Montano, A.; Aparicio-Puerta, E.; Hackenberg, M.; Carmona-Saez, P. GeneCodis 4: Expanding the modular enrichment analysis to regulatory elements. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jaffe, E.F.; Lejtenyi, M.C.; Noya, F.J.; Mazer, B.D. Secondary hypogammaglobulinemia. Immunol. Allergy Clin. N. Am. 2001, 21, 141–163. [Google Scholar] [CrossRef]
- Tsirlin, A.; Oo, Y.; Sharma, R.; Kansara, A.; Gliwa, A.; Banerji, M.A. Heochromocytoma: A review. Maturitas 2014, 77, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Brunzell, J.D. Hypertriglyceridemia. N. Engl. J. Med. 2007, 357, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.G.; Johnson, T.L.; Marson, A.G.; Chadwick, D.W. Prediction of risk of seizure recurrence after a single seizure and early epilepsy: Further results from the MESS trial. Lancet Neurol. 2006, 5, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Mechaussier, S.; Almoallem, B.; Zeitz, C.; Van Schil, K.; Jeddawi, L.; Van Dorpe, J.; Rey, A.D.; Condroyer, C.; Pelle, O.; Polak, M.; et al. Loss of function of RIMS2 causes a syndromic congenital cone-rod synaptic disease with neurodevelopmental and pancreatic involvement. Am. J. Hum. Genet. 2020, 106, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.Y.R.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-onset familial Alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef] [PubMed]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31, S262–S268. [Google Scholar] [CrossRef] [PubMed]
- De Vries, L.; Kauschansky, A.; Shohat, M.; Phillip, M. Familial central precocious puberty suggests autosomal dominant inheritance. J. Clin. Endocrinol. Metab. 2004, 89, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Magid-Bernstein, J.; Girard, R.; Polster, S.; Srinath, A.; Romanos, S.; Awad, I.A.; Sansing, L.H. Cerebral hemorrhage: Pathophysiology, treatment, and future directions. Circ. Res. 2022, 130, 1204–1229. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.L.; Aliverti, A. Physiology of respiratory disturbances in muscular dystrophies. Breathe 2016, 12, 318–327. [Google Scholar] [CrossRef]
- Wagner, R.; Tse, W.H.; Gosemann, J.H.; Lacher, M.; Keijzer, R. Prenatal maternal biomarkers for the early diagnosis of congenital malformations: A review. Pediatr. Res. 2019, 86, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic pulmonary fibrosis (IPF): An overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | No. of Samples in Population | Lines of Input | Processed Variants | Novel/Existing Variants | Overlapped Genes | Overlapped Transcripts |
|---|---|---|---|---|---|---|
| HC_Male | 50 | 18,191,327 | 18,191,327 | 168,623 (0.9%)/18,022,704 (99.1%) | 61,987 | 250,277 |
| HC_Female | 50 | 18,387,115 | 18,387,115 | 182,458 (1.0%)/18,204,657 (99.0%) | 61,663 | 249,831 |
| Pro_Gen_Male | 61 | 17,763,968 | 17,763,968 | 179,814 (1.0%)/17,584,154 (99.0%) | 62,009 | 250,316 |
| Pro_Gen_Female | 85 | 19,371,326 | 19,371,326 | 228,369 (1.2%)/19,142,957 (98.8%) | 61,655 | 249,834 |
| Pro_RBD_Male | 31 | 12,264,624 | 12,264,624 | 63,331 (0.5%)/12,201,293 (99.5%) | 61,898 | 250,173 |
| Pro_RBD_Female | 6 | 9,568,588 | 9,568,588 | 40,093 (0.4%)/9,528,495 (99.6%) | 61,542 | 249,619 |
| Pro_Hypo_Male | 14 | 12,081,647 | 12,081,647 | 69,229 (0.6%)/12,012,418 (99.4%) | 61,924 | 250,185 |
| Pro_Hypo_Female | 7 | 9,942,790 | 9,942,790 | 29,510 (0.3%)/9,913,280 (99.7%) | 61,565 | 249,627 |
| Population | Disease | Number of Genes | Dataset |
|---|---|---|---|
| HC_Male | Acquired Hypogammaglobulinemia | 11 | DisGeNET |
| Hyperinsulinemia | 120 | HPO | |
| HC_Female | Dermatologic Disorders | 75 | DisGeNET |
| Autosomal Dominant Inheritance | 1828 | HPO | |
| Pro_Gen_Male | Pheochromocytoma | 19 | DisGeNET |
| Cerebral Hemorrhage | 62 | HPO | |
| Pro_Gen_Female | Single Seizure | 101 | DisGeNET |
| Autosomal Dominant Inheritance | 1828 | HPO | |
| Pro_RBD_Male | Cone–Rod Synaptic Disorder (CRSD) | 13 | DisGeNET |
| Respiratory Insufficiency Due to Muscle Weakness | 79 | HPO | |
| Pro_RBD_Female | Autosomal Recessive Primary Microcephaly | 22 | DisGeNET |
| Autosomal Dominant Inheritance | 1828 | HPO | |
| Pro_Hypo_Male | Arthritis; Adjuvant-Induced | 40 | DisGeNET |
| Prenatal Maternal Abnormality | 23 | HPO | |
| Pro_Hypo_Female | Familial Alzheimer’s Disease (FAD) | 99 | DisGeNET |
| Autosomal Dominant Inheritance | 1828 | HPO |
| Population | Pro_Gen_Male Gene Count | Pro_Gen_Female Gene Count | Pro_RBD_Male Gene Count | Pro_RBD_Female Gene Count | Pro_Hypo_Male Gene Count | Pro_Hypo_Female Gene Count | |
|---|---|---|---|---|---|---|---|
| Gene Name | |||||||
| MTF2 | 2985 | 3458 | 3065 | 1397 | 2035 | 1606 | |
| ADD1 | 9095 | 10,457 | 5535 | 3471 | 4404 | 3722 | |
| PIK3CA | 2958 | 3072 | 1874 | 1390 | 1606 | 1830 | |
| SYBU | 13,897 | 15,607 | 7991 | 5943 | 8012 | 6030 | |
| IRS2 | 237 | 291 | 192 | 138 | 210 | 147 | |
| USP8 | 5997 | 6524 | 4432 | 3358 | 4953 | 1953 | |
| PIGL | 8277 | 9021 | 4350 | 2819 | 3902 | 2923 | |
| FASN | 1184 | 1224 | 691 | 552 | 775 | 593 | |
| MYLK2 | 265 | 296 | 183 | 132 | 111 | 107 | |
| USP25 | 4081 | 4952 | 2672 | 753 | 2796 | 534 | |
| EP300 | 3146 | 3510 | 1704 | 1256 | 1790 | 1398 | |
| PPP6R2 | 5652 | 6393 | 3090 | 2693 | 3723 | 2561 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badawy, M.T.; Salama, A.A.; Salama, M. Novel Variants Linked to the Prodromal Stage of Parkinson’s Disease (PD) Patients. Diagnostics 2024, 14, 929. https://doi.org/10.3390/diagnostics14090929
Badawy MT, Salama AA, Salama M. Novel Variants Linked to the Prodromal Stage of Parkinson’s Disease (PD) Patients. Diagnostics. 2024; 14(9):929. https://doi.org/10.3390/diagnostics14090929
Chicago/Turabian StyleBadawy, Marwa T., Aya A. Salama, and Mohamed Salama. 2024. "Novel Variants Linked to the Prodromal Stage of Parkinson’s Disease (PD) Patients" Diagnostics 14, no. 9: 929. https://doi.org/10.3390/diagnostics14090929
APA StyleBadawy, M. T., Salama, A. A., & Salama, M. (2024). Novel Variants Linked to the Prodromal Stage of Parkinson’s Disease (PD) Patients. Diagnostics, 14(9), 929. https://doi.org/10.3390/diagnostics14090929