


Progressive Age-Associated Blood–Brain Barrier Leak/Dysfunction-Nexus of Neurodegenerative Disease Using MRI Markers to Identify Preclinical Disease and Potential New Targets for Future Treatments


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Normal BBB Physiology and Regulation
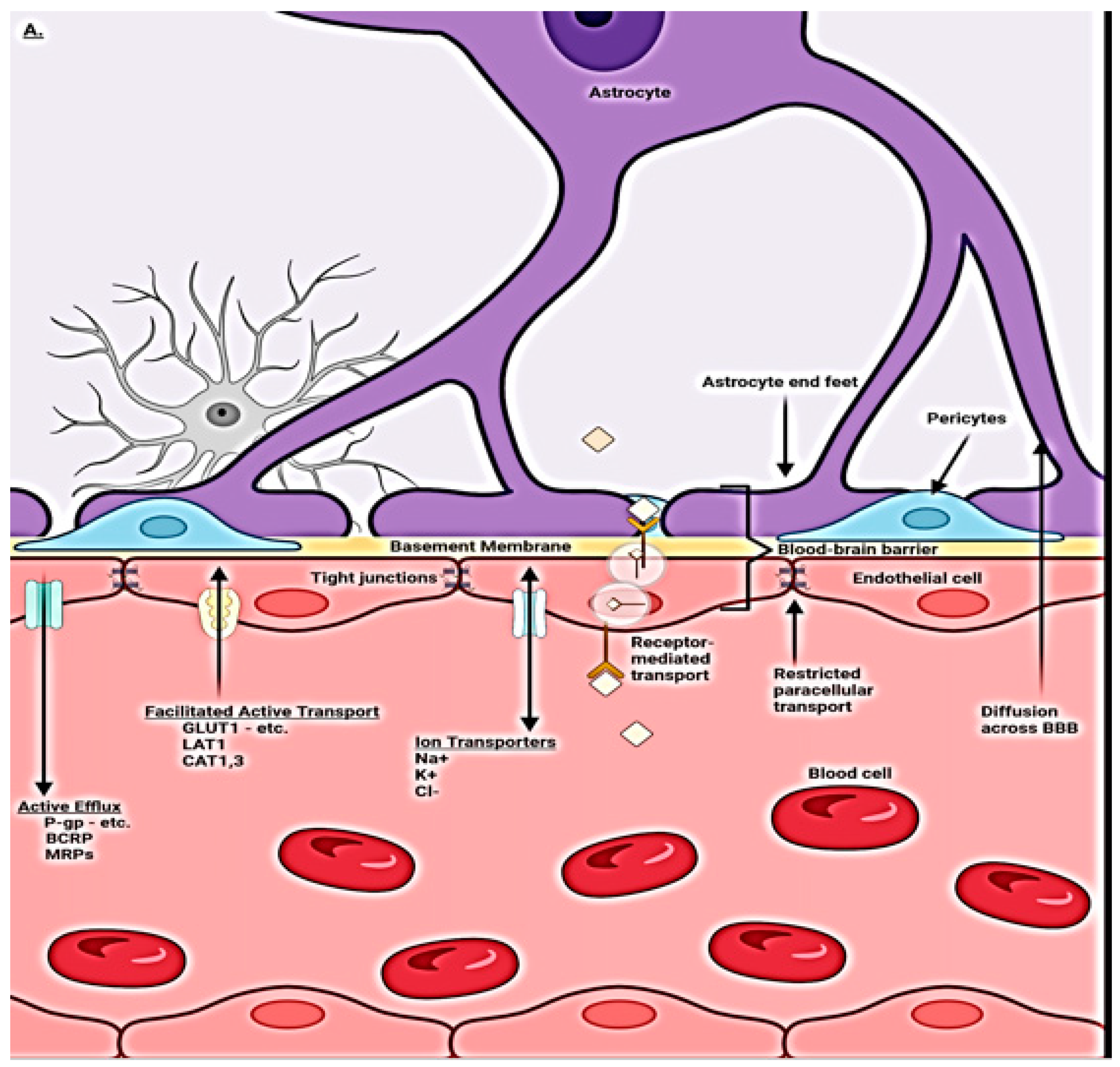
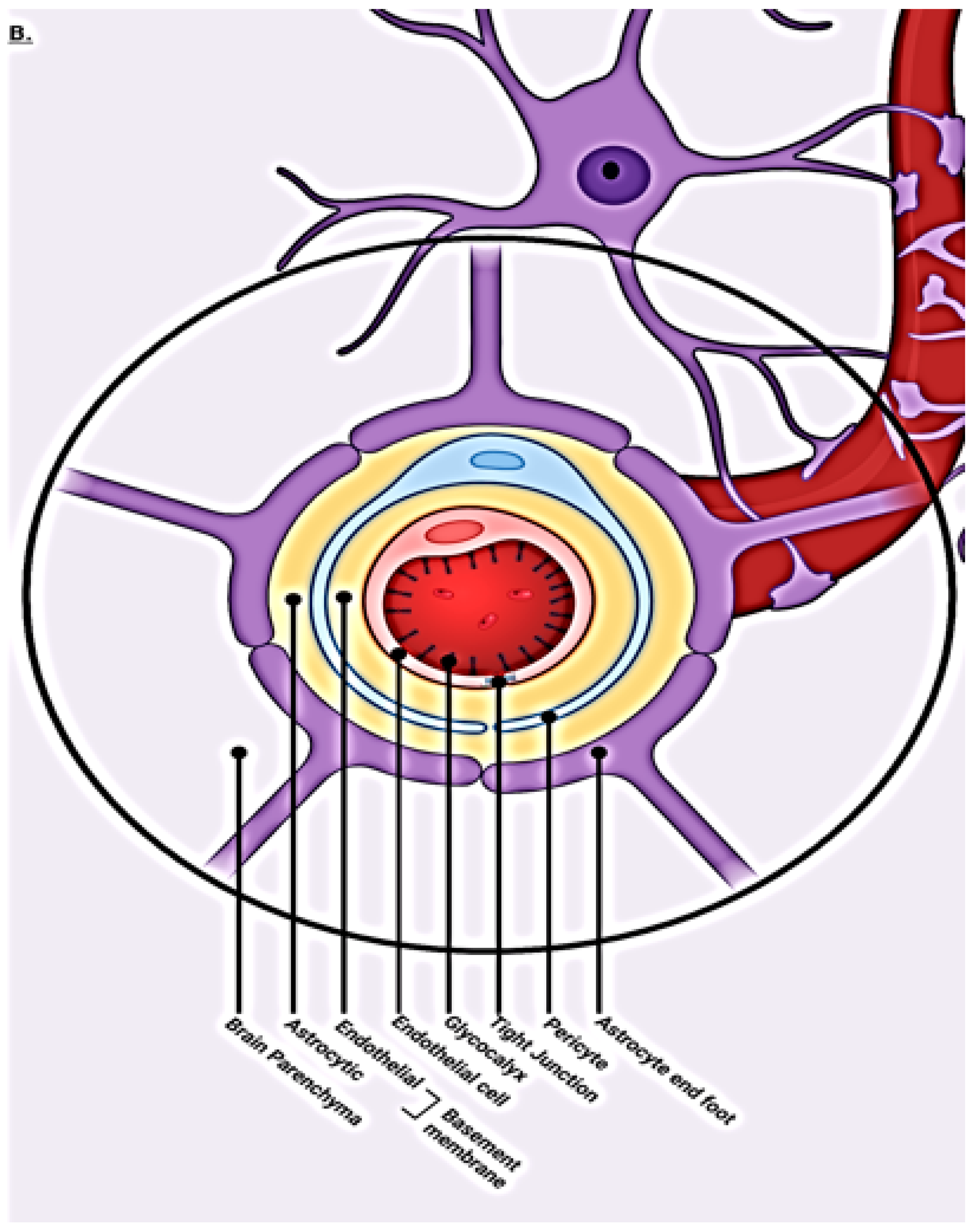
1.2. Blood–Brain Barrier Cellular Makeup and Their Function
1.3. Capillary Endothelial Cells
1.4. Pericytes
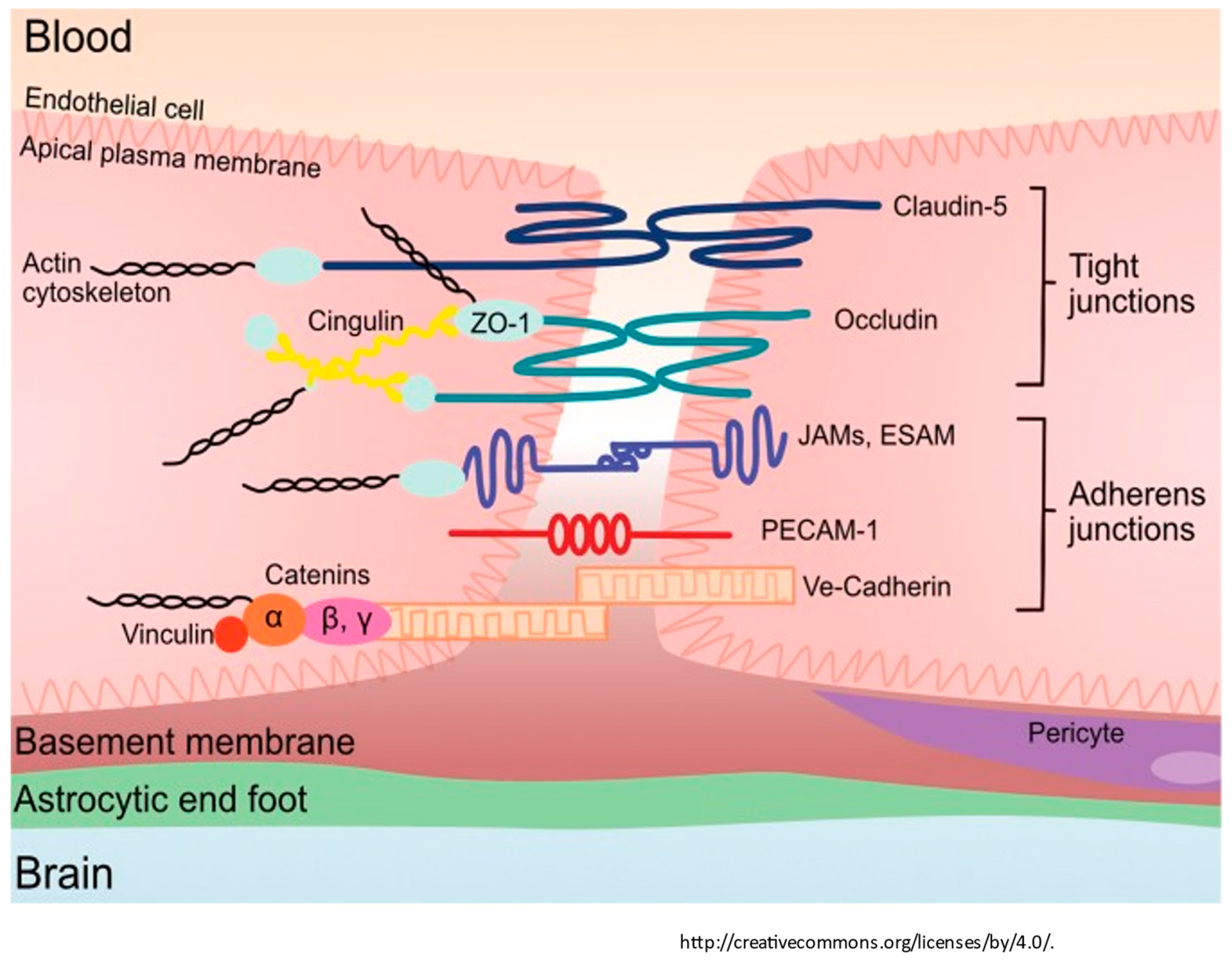
1.5. Tight Junctions
1.6. Astrocytes
1.7. Microglia
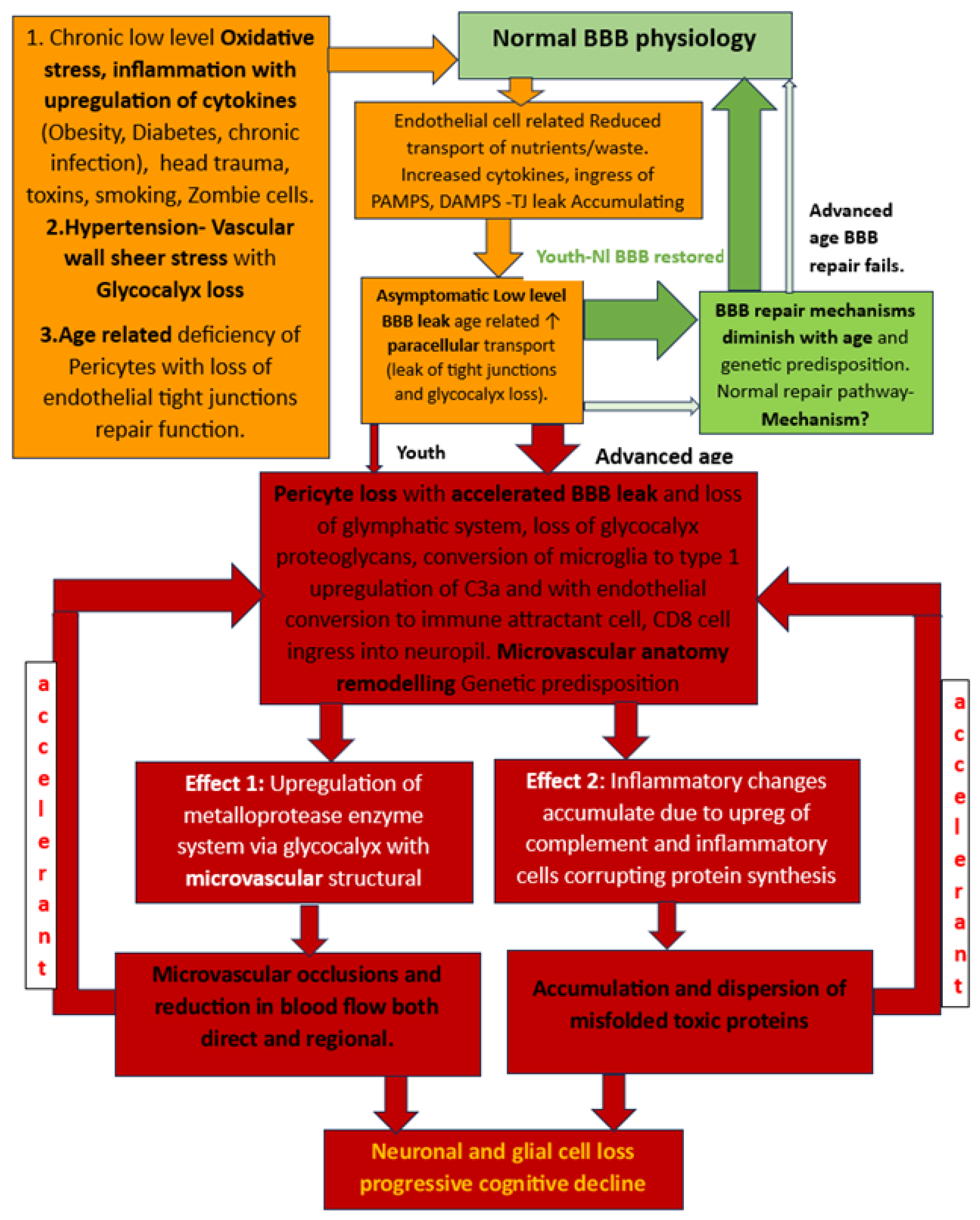
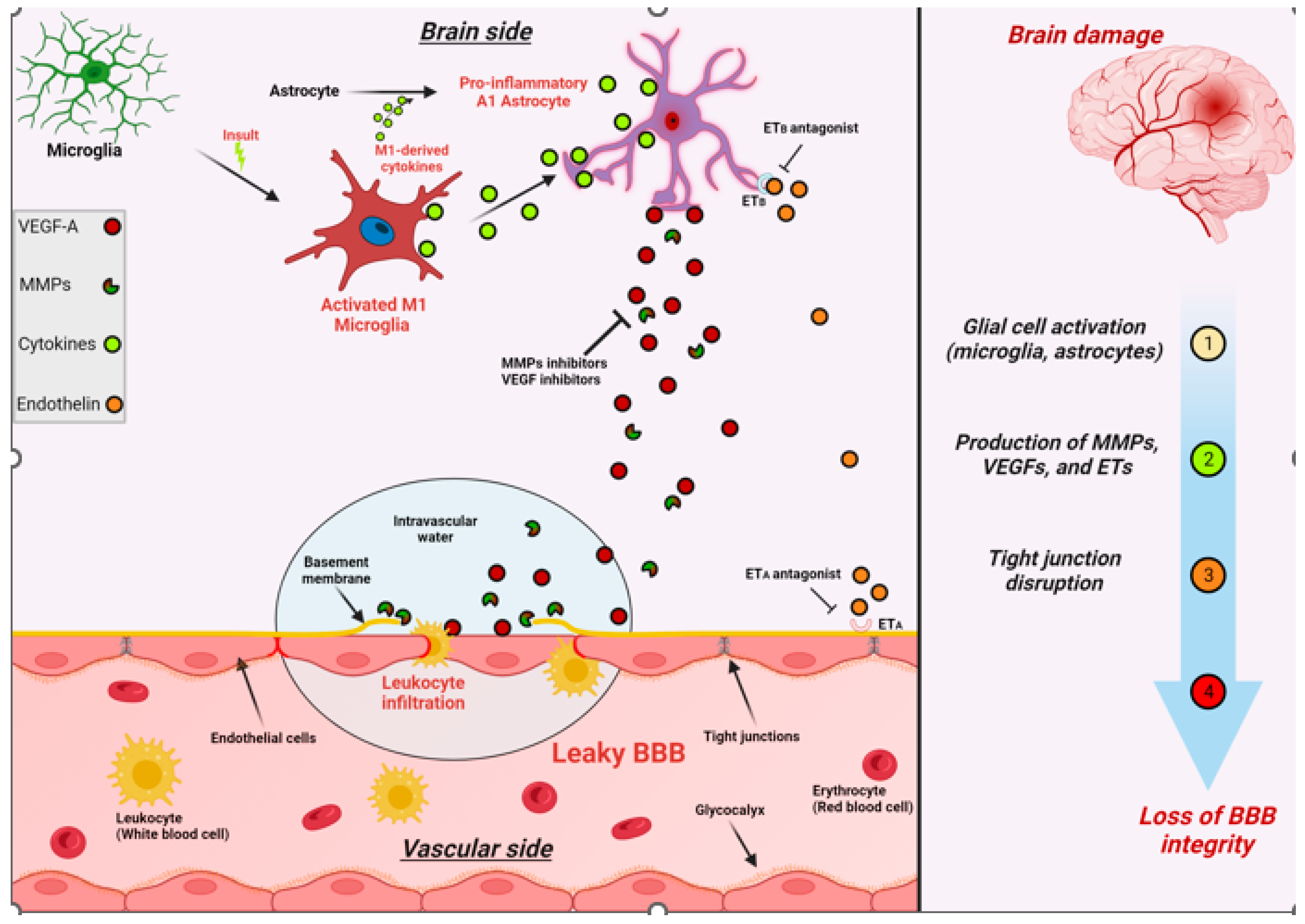
1.8. BBB Dysfunction
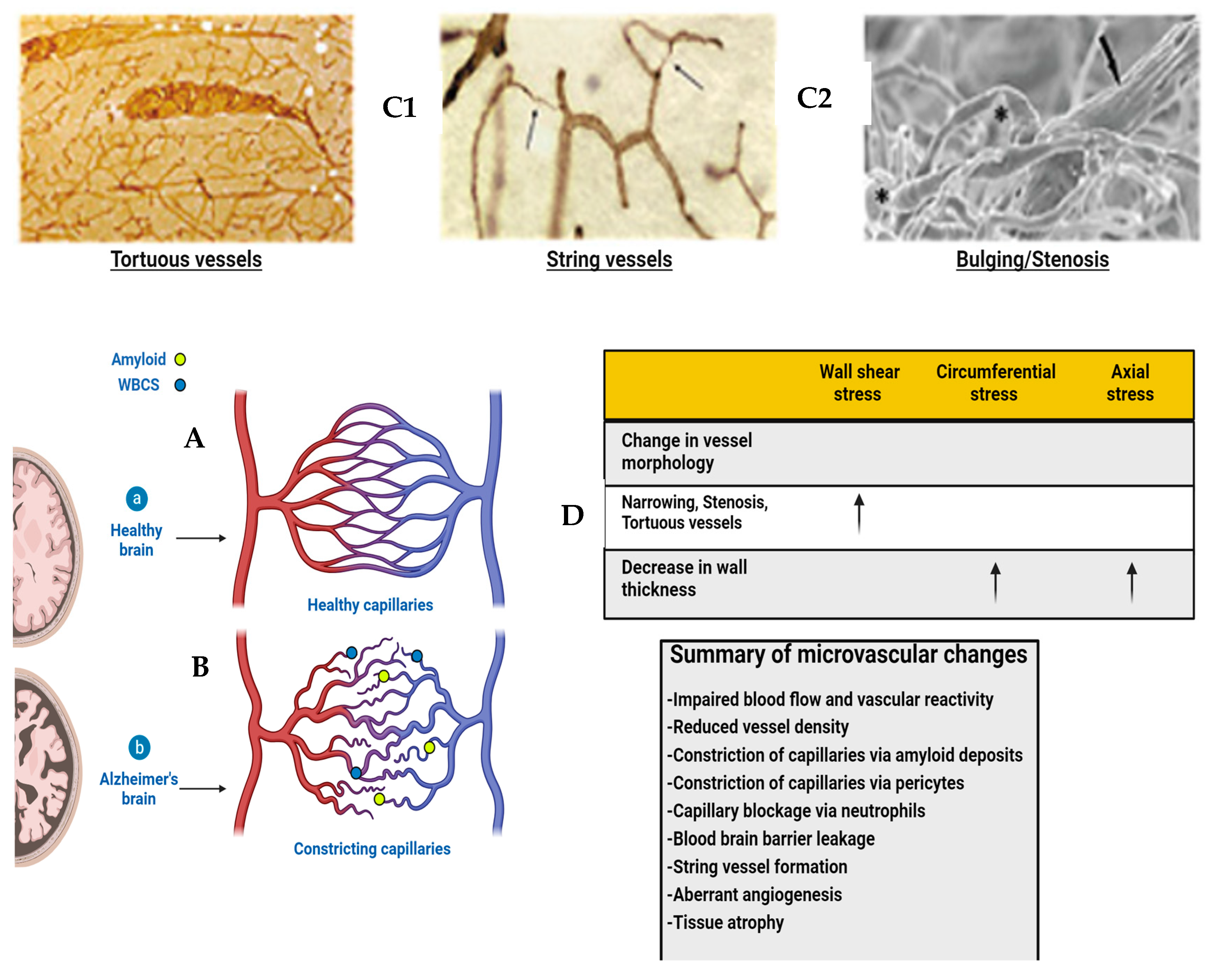
2. Altered Fluid Dynamic
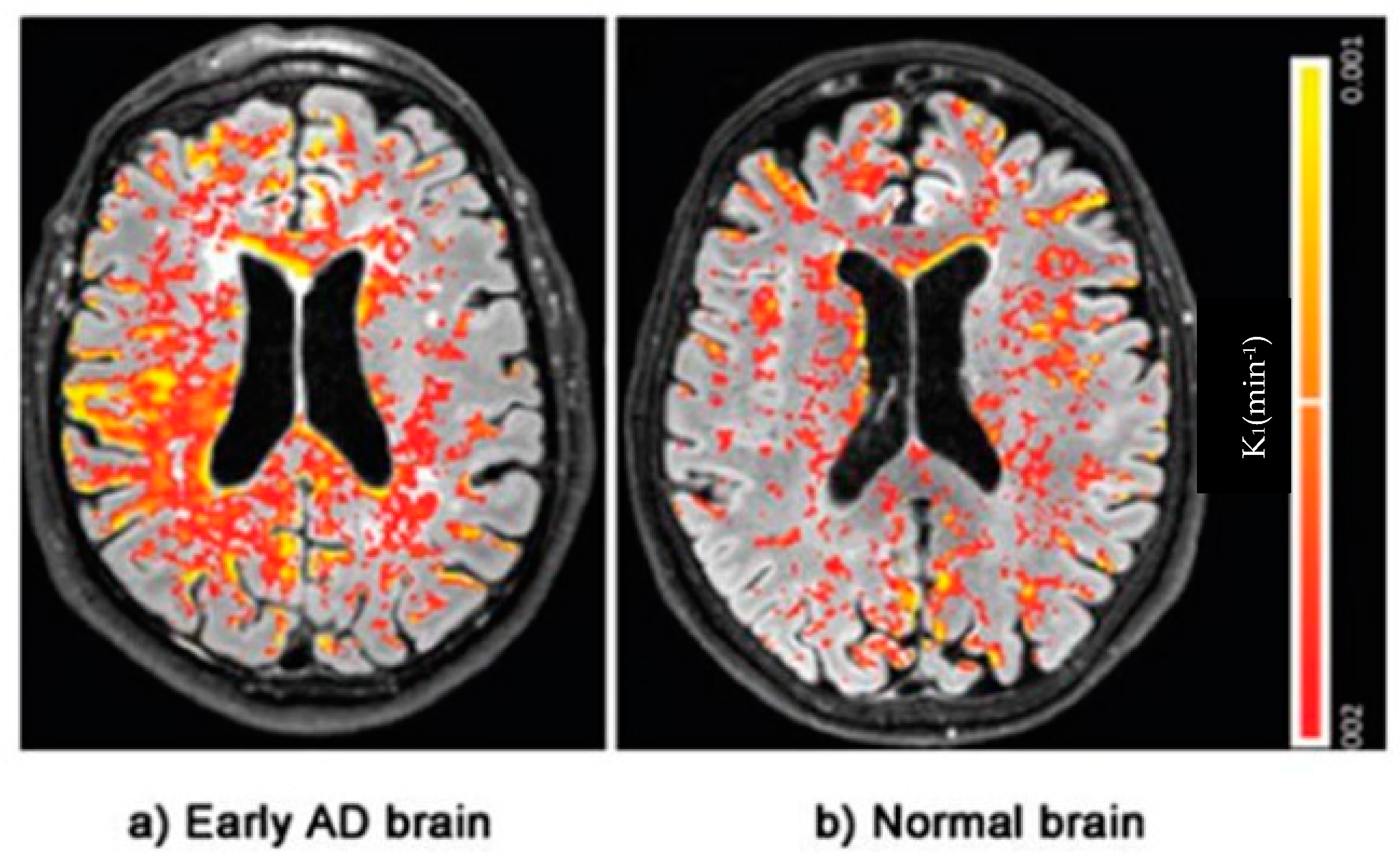
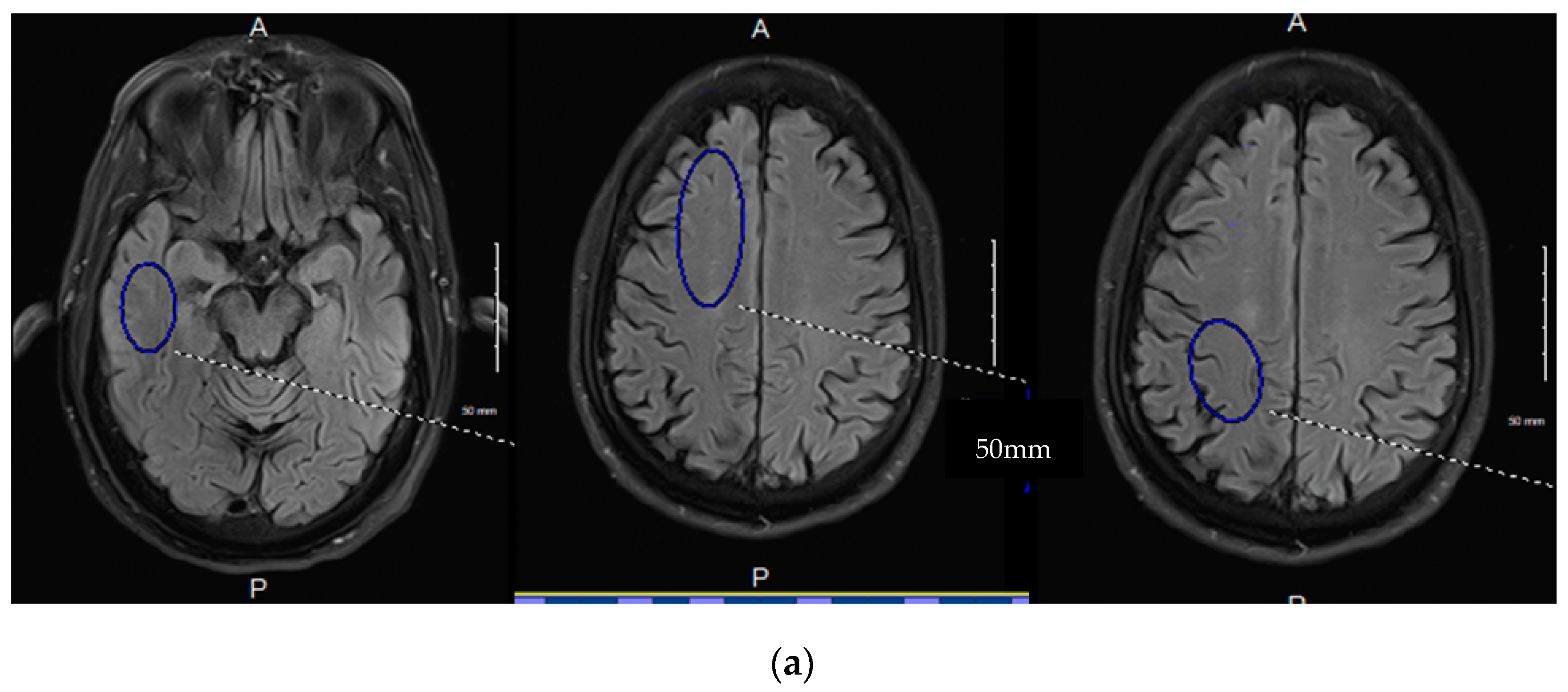
3. Early Identification of BBB Dysfunction
4. Current Methods of Risk Reduction
5. Conclusions—Where Do We Go from Here?
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ebell, M.H.; Barry, H.C.; Baduni, K.; Grasso, G. Clinically Important Benefits and Harms of Monoclonal Antibodies Targeting Amyloid for the Treatment of Alzheimer Disease: A Systematic Review and Meta-Analysis. Ann. Fam. Med. 2024, 22, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Smith, J.; Donohue, M.C.; Delmar, P.; Abbas, R.; Salloway, S.; Wojtowicz, J. Two Phase 3 Trials of Gantenerumab in Early Alzheimer’s Disease. Engl. J. Med. 2023, 389, 1862–1876. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wong, L.-W.; Su, Y.; Huang, X.; Wang, N.; Chen, H.; Yi, C. Blood-brain barrier integrity in the pathogenesis of Alzheimer’s disease. Front. Neuroendocrinol. 2020, 59, 100857. [Google Scholar] [CrossRef] [PubMed]
- Barisano, G.; Montagne, A.; Kisler, K.; Schneider, J.A.; Wardlaw, J.M.; Zlokovic, B.V. Blood–brain barrier link to human cognitive impairment and Alzheimer’s disease. Nat. Cardiovasc. Res. 2022, 1, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Hussain, B.; Cheng, F.; Junlei, C. Blood–brain barrier breakdown: An emerging biomarker of cognitive impairment in normal aging and dementia. Front. Neurosci. 2021, 15, 688090. [Google Scholar] [CrossRef] [PubMed]
- Nehra, G.; Bjoern, B.; Hartz, A.M. Blood-brain barrier leakage in Alzheimer’s disease: From discovery to clinical relevance. Pharmacol. Ther. 2022, 234, 108119. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, A.V.; Situ, M.; Citalan-Madrid, A.F.; Stamatovic, S.M.; Xiang, J.; Keep, R.F. Blood-brain barrier dysfunction in normal aging and neurodegeneration: Mechanisms, impact, and treatments. Stroke 2023, 54, 661–672. [Google Scholar] [CrossRef]
- Selkoe, D.J. Treatments for Alzheimer’s disease emerge. Science 2021, 373, 624–626. [Google Scholar] [CrossRef]
- Castro Dias, M.; Mapunda, J.A.; Vladymyrov, M.; Engelhardt, B. Structure and junctional complexes of endothelial, epithelial and glial brain barriers. Int. J. Mol. Sci. 2019, 20, 5372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Q.; He, C.; Fan, D.; Zhu, Y.; Zang, F.; Tan, C. Altered regional cerebral blood flow and brain function across the Alzheimer’s disease spectrum: A potential biomarker. Front. Aging Neurosci. 2021, 13, 630382. [Google Scholar] [CrossRef] [PubMed]
- Thrippleton, M.J.; Backes, W.H.; Sourbron, S.; Ingrisch, M.; van Osch, M.J.P.; Dichgans, M.; Fazekas, F. Quantifying blood-brain barrier leakage in small vessel disease: Review and consensus recommendations. Alzheimer’s Dement. 2019, 15, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Hu, H.; Liu, C.; Sun, N.; Duan, C. Methods used for the measurement of blood-brain barrier integrity. Metab. Brain Dis. 2021, 36, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.B.; Janelidze, S.; Smith, R.; Mattsson-Carlgren, N.; Palmqvist, S.; Teunissen, C.E.; Zetterberg, H. Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain 2021, 144, 3505–3516. [Google Scholar] [CrossRef]
- Kadry, H.; Behnam, N.; Luca, C. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Zhao, L.; Tannenbaum, A.; Bakker, E.N.T.; Benveniste, H. Physiology of glymphatic solute transport and waste clearance from the brain. Physiology 2022, 37, 349–362. [Google Scholar] [CrossRef]
- Preston, J.E.; Abbott, N.J.; Begley, D.J. Transcytosis of macromolecules at the blood–brain barrier. Adv. Pharmacol. 2014, 71, 147–163. [Google Scholar]
- van Leeuwen, E.; Hampton, M.B.; Smyth, L.C.D. Redox signaling and regulation of the blood-brain barrier. Int. J. Biochem. Cell Biol. 2020, 125, 105794. [Google Scholar] [CrossRef]
- Proulx, S.T. Cerebrospinal fluid outflow: A review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics. Cell. Mol. Life Sci. 2021, 78, 2429–2457. [Google Scholar] [CrossRef]
- Griffith, J.I.; Rathi, S.; Zhang, W.; Zhang, W.; Drewes, L.R.; Sarkaria, J.N.; Elmquist, W.F. Addressing BBB heterogeneity: A new paradigm for drug delivery to brain tumors. Pharmaceutics 2020, 12, 1205. [Google Scholar] [CrossRef]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- Hudson, N.; Matthew, C. Tight junctions of the neurovascular unit. Front. Mol. Neurosci. 2021, 14, 752781. [Google Scholar] [CrossRef]
- Engelhardt, B.; Stefan, L. Novel insights into the development and maintenance of the blood–brain barrier. Cell Tissue Res. 2014, 355, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’Driscoll, C.M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Wallerstein, A.; Abdul-Muneer, P. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 2019, 317, 260–270. [Google Scholar] [CrossRef]
- Brassard, P.; Labrecque, L.; Smirl, J.D.; Tymko, M.M.; Caldwell, H.G.; Hoiland, R.L.; Lucas, S.J.E.; Denault, A.Y.; Couture, E.J.; Ainslie, P.N. Losing the dogmatic view of cerebral autoregulation. Physiol. Rep. 2021, 9, e14982. [Google Scholar] [CrossRef]
- Boyé, K.; Geraldo, L.H.; Furtado, J.; Pibouin-Fragner, L.; Poulet, M.; Kim, D.; Nelson, B. Endothelial Unc5B controls blood-brain barrier integrity. Nat. Commun. 2022, 13, 1169. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Ahn, S.J.; Lane, D.; Faraco, G.; Garcia-Bonilla, L.; Racchumi, G.; Poon, C. Endothelium-macrophage crosstalk mediates blood-brain barrier dysfunction in hypertension. Hypertension 2020, 76, 795–807. [Google Scholar] [CrossRef]
- Roudnicky, F.; Zhang, J.D.; Kim, B.K.; Pandya, N.J.; Lan, Y.; Sach-Peltason, L.; Ragelle, H. Inducers of the endothelial cell barrier identified through chemogenomic screening in genome-edited hPSC-endothelial cells. Proc. Natl. Acad. Sci. USA 2020, 117, 19854–19865. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood–brain barrier in health and disease: Important unanswered questions. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Chen, M.; Zheng, J.; Li, X.; Zhang, X. The Role of Heparin and Glycocalyx in Blood–Brain Barrier Dysfunction. Front. Immunol. 2021, 12, 754141. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Per, N.; Christer, B. Emerging links between cerebrovascular and neurodegenerative diseases—A special role for pericytes. EMBO Rep. 2019, 20, e48070. [Google Scholar] [CrossRef]
- Zhao, N.; Guo, Z.; Pessell, A.F.; Searson, P.C. The influence of physiological and pathological perturbations on blood-brain barrier function. Front. Neurosci. 2023, 17, 1289894. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood–brain barrier. Nat. Aging 2021, 1, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Verheggen, I.C.M.; de Jong, J.J.A.; van Boxtel, M.P.J.; Gronenschild, E.H.B.; Palm, W.M.; Postma, A.A.; Jansen, J.F.A.; Verhey, F.R.J.; Backes, W.H. Increase in blood–brain barrier leakage in healthy, older adults. Geroscience 2020, 42, 1183–1193. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Mestre, H.; Yuki, M.; Maiken, N. The brain’s glymphatic system: Current controversies. Trends Neurosci. 2020, 43, 458–466. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Thomas, P.D. Regulation of blood–brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Early role of vascular dysregulation on late-onset alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Cash, A.; Michelle, H.T. Mechanisms of blood–brain barrier dysfunction in traumatic brain injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Katsi, V.; Marketou, M.; Maragkoudakis, S.; Didagelos, M.; Charalambous, G.; Parthenakis, F.; Tsioufis, C.; Tousoulis, D. Blood–brain barrier dysfunction: The undervalued frontier of hypertension. Hum. Hypertens. 2020, 34, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.J.E.; Tzeng, Y.C.; Galvin, S.D.; Thomas, K.N.; Ogoh, S.; Ainslie, P.N. Influence of changes in blood pressure on cerebral perfusion and oxygenation. Hypertension 2010, 55, 698–705. [Google Scholar] [CrossRef]
- Klohs, J. An integrated view on vascular dysfunction in Alzheimer’s disease. Neurodegener. Dis. 2020, 19, 109–127. [Google Scholar] [CrossRef]
- Vázquez-Rosa, E.; Shin, M.-K.; Dhar, M.; Chaubey, K.; Cintrón-Pérez, C.J.; Tang, X.; Liao, X. P7C3-A20 treatment one year after TBI in mice repairs the blood–brain barrier, arrests chronic neurodegeneration, and restores cognition. Proc. Natl. Acad. Sci. USA 2020, 117, 27667–27675. [Google Scholar] [CrossRef]
- Joseph, C.R.; Lim, J.K.; Grohol, B.N.; Zivcevska, M.; Lencke, J.; Rich, E.D.; Arrasmith, C.J. Identifying delay in glymphatic clearance of labeled protons post-acute head trauma utilizing 3D ASL MRI (arterial spin labeling): A pilot study. Sci. Rep. 2024, 14, 6188. [Google Scholar] [CrossRef]
- Vigasova, D.; Nemergut, M.; Liskova, B.; Damborsky, J. Multi-pathogen infections and Alzheimer’s disease. Microb. Cell Factories 2021, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Jessy, J.A. Complement and blood–brain barrier integrity. Mol. Immunol. 2014, 61, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, H.; Luo, Y.; Tan, X. Correlation of serum complement factor 5a level with inflammatory response and cognitive function in patients with Alzheimer’s disease of different severity. BMC Neurol. 2023, 23, 319. [Google Scholar]
- Eadon, M.T.; Jacob, A.; Cunningham, P.N.; Quigg, R.J.; Garcia, J.G.; Alexander, J.J. Transcriptional profiling reveals that C5a alters miRNA in brain endothelial cells. Immunology 2014, 143, 363–373. [Google Scholar] [CrossRef]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Köhl, J.; Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Uday, K.; Abhishek, S. Complement system in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 13647. [Google Scholar] [CrossRef] [PubMed]
- Krance, S.H.; Wu, C.-Y.; Zou, Y.; Mao, H.; Toufighi, S.; He, X.; Pakosh, M.; Swardfager, W. The complement cascade in Alzheimer’s disease: A systematic review and meta-analysis. Mol. Psychiatry 2021, 26, 5532–5541. [Google Scholar] [CrossRef]
- Elschot, E.P.; Backes, W.H.; Postma, A.A.; van Oostenbrugge, R.J.; Staals, J.; Rouhl, R.P.W.; Jansen, J.F.A. A comprehensive view on MRI techniques for imaging blood-brain barrier integrity. Investig. Radiol. 2021, 56, 10–19. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Chang, J.; Mancuso, M.R.; Maier, C.; Liang, X.; Yuki, K.; Yang, L.U.; Kwong, J.W. Gpr124 is essential for blood–brain barrier integrity in central nervous system disease. Nat. Med. 2017, 23, 450–460. [Google Scholar] [CrossRef]
- Travagli, R.; Alberto, K.N.; Camilleri, M. Parkinson disease and the gut: New insights into pathogenesis and clinical relevance. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 673–685. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Liu, K.; Zhang, H.-L. Microbiota-gut-brain axis and Alzheimer’s disease: Implications of the blood-brain barrier as an intervention target. Mech. Ageing Dev. 2021, 199, 111560. [Google Scholar] [CrossRef] [PubMed]
- Ahumada-Castro, U.; Puebla-Huerta, A.; Cuevas-Espinoza, V.; Lovy, A.; Cardenas, J.C. Keeping zombies alive: The ER-mitochondria Ca2+ transfer in cellular senescence. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 119099. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-brain barrier dysfunction amplifies the development of neuroinflammation: Understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef] [PubMed]
- Yoshiura, T.; Hiwatashi, A.; Yamashita, K.; Ohyagi, Y.; Monji, A.; Takayama, Y.; Nagao, E.; Kamano, H.; Noguchi, T.; Honda, H. Simultaneous measurement of arterial transit time, arterial blood volume, and cerebral blood flow using arterial spin-labeling in patients with Alzheimer disease. Am. Neuroradiol. 2009, 30, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Binnewijzend, M.A.A.; Benedictus, M.R.; Kuijer, J.P.A.; van der Flier, W.M.; Teunissen, C.E.; Prins, N.D.; Wattjes, M.P.; van Berckel, B.N.M.; Scheltens, P.; Barkhof, F. Cerebral perfusion in the predementia stages of Alzheimer’s disease. Eur. Radiol. 2016, 26, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Cui, B.; Han, Y.; He, Y.; Wang, Z. Disrupted regional cerebral blood flow, functional activity and connectivity in Alzheimer’s disease: A combined ASL perfusion and resting state fMRI study. Front. Neurosci. 2019, 13, 738. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, N.; Barbui, C.; Anstey, K.J.; Kivipelto, M.; Barbera, M.; Zheng, L.; Kulmala, J.; Stephen, R.; Joanette, Y. Reducing the risk of cognitive decline and dementia: WHO recommendations. Front. Neurol. 2022, 12, 765584. [Google Scholar] [CrossRef] [PubMed]
- Steinman, J.; Sun, H.-S.; Feng, Z.-P. Microvascular alterations in Alzheimer’s disease. Front. Cell. Neurosci. 2021, 14, 618986. [Google Scholar] [CrossRef]
- Meyer, E.P.; Ulmann-Schuler, A.; Staufenbiel, M.; Krucker, T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 3587–3592. [Google Scholar] [CrossRef]
- Dorr, A.; Sahota, B.; Chinta, L.V.; Brown, M.E.; Lai, A.Y.; Ma, K.; Hawkes, C.A.; McLaurin, J.; Stefanovic, B. Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer’s disease. Brain 2012, 135, 3039–3050. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Archie, S.R.; Abdullah, A.S.; Luca, C. Blood-brain barrier dysfunction in CNS disorders and putative therapeutic targets: An overview. Pharmaceutics 2021, 13, 1779. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Jäkel, L.; De Kort, A.M.; Klijn, C.J.M.; Schreuder, F.H.B.; Verbeek, M.M. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimer’s Dement. 2022, 18, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Fazlollahi, A.; Calamante, F.; Liang, X.; Bourgeat, P.; Raniga, P.; Dore, V.; Fripp, J. Increased cerebral blood flow with increased amyloid burden in the preclinical phase of alzheimer’s disease. J. Magn. Reson. Imaging 2020, 51, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; Van Osch, M.J.P.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Nation, D.A.; Kisler, K.; Toga, A.W.; Zlokovic, B.V. Imaging subtle leaks in the blood–brain barrier in the aging human brain: Potential pitfalls, challenges, and possible solutions. GeroScience 2022, 44, 1339–1351. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.R.; Benhatzel, C.M.; Stern, L.J.; Hopper, O.M.; Lockwood, M.D. Pilot study utilizing MRI 3D TGSE PASL (arterial spin labeling) differentiating clearance rates of labeled protons in the CNS of patients with early Alzheimer disease from normal subjects. Magn. Reson. Mater. Phys. Biol. Med. 2020, 33, 559–568. [Google Scholar] [CrossRef]
- Joseph, C.R.; Kreilach, A.; Reyna, V.A.; Kepler, T.A.; Taylor, B.V.; Kang, J.; McCorkle, D.; Rider, N.L. Utilizing Reduced Labeled Proton Clearance to Identify Preclinical Alzheimer Disease with 3D ASL MRI. Case Rep. Neurol. 2023, 15, 177. [Google Scholar] [CrossRef]
- Mahapatra, M.K.; Muthukumar, K.; Biswa, M.S. Therapeutic potential of semaglutide, a newer GLP-1 receptor agonist, in abating obesity, non-alcoholic steatohepatitis and neurodegenerative diseases: A narrative review. Pharm. Res. 2022, 39, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, M.M.; Garbarino, V.R.; Zilli, E.M.; Petersen, R.C.; Kirkland, J.L.; Tchkonia, T.; Musi, N.; Seshadri, S.; Craft, S.; Orr, M.E. Senolytic therapy to modulate the progression of Alzheimer’s disease (SToMP-AD): A pilot clinical trial. J. Prev. Alzheimer’s Dis. 2022, 9, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tamar, T.; James, L.K. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, C.R. Progressive Age-Associated Blood–Brain Barrier Leak/Dysfunction-Nexus of Neurodegenerative Disease Using MRI Markers to Identify Preclinical Disease and Potential New Targets for Future Treatments. Diagnostics 2024, 14, 726. https://doi.org/10.3390/diagnostics14070726
Joseph CR. Progressive Age-Associated Blood–Brain Barrier Leak/Dysfunction-Nexus of Neurodegenerative Disease Using MRI Markers to Identify Preclinical Disease and Potential New Targets for Future Treatments. Diagnostics. 2024; 14(7):726. https://doi.org/10.3390/diagnostics14070726
Chicago/Turabian StyleJoseph, Charles R. 2024. "Progressive Age-Associated Blood–Brain Barrier Leak/Dysfunction-Nexus of Neurodegenerative Disease Using MRI Markers to Identify Preclinical Disease and Potential New Targets for Future Treatments" Diagnostics 14, no. 7: 726. https://doi.org/10.3390/diagnostics14070726
APA StyleJoseph, C. R. (2024). Progressive Age-Associated Blood–Brain Barrier Leak/Dysfunction-Nexus of Neurodegenerative Disease Using MRI Markers to Identify Preclinical Disease and Potential New Targets for Future Treatments. Diagnostics, 14(7), 726. https://doi.org/10.3390/diagnostics14070726