
Cutaneous Intravascular Hematolymphoid Entities: A Review
 ,
,
Abstract
1. Introduction
2. Intravascular B-Cell Lymphoma
2.1. Clinical Presentation
2.1.1. Epidemiology
2.1.2. Clinical Features
2.1.3. Diagnostics
2.1.4. Treatment and Outcomes
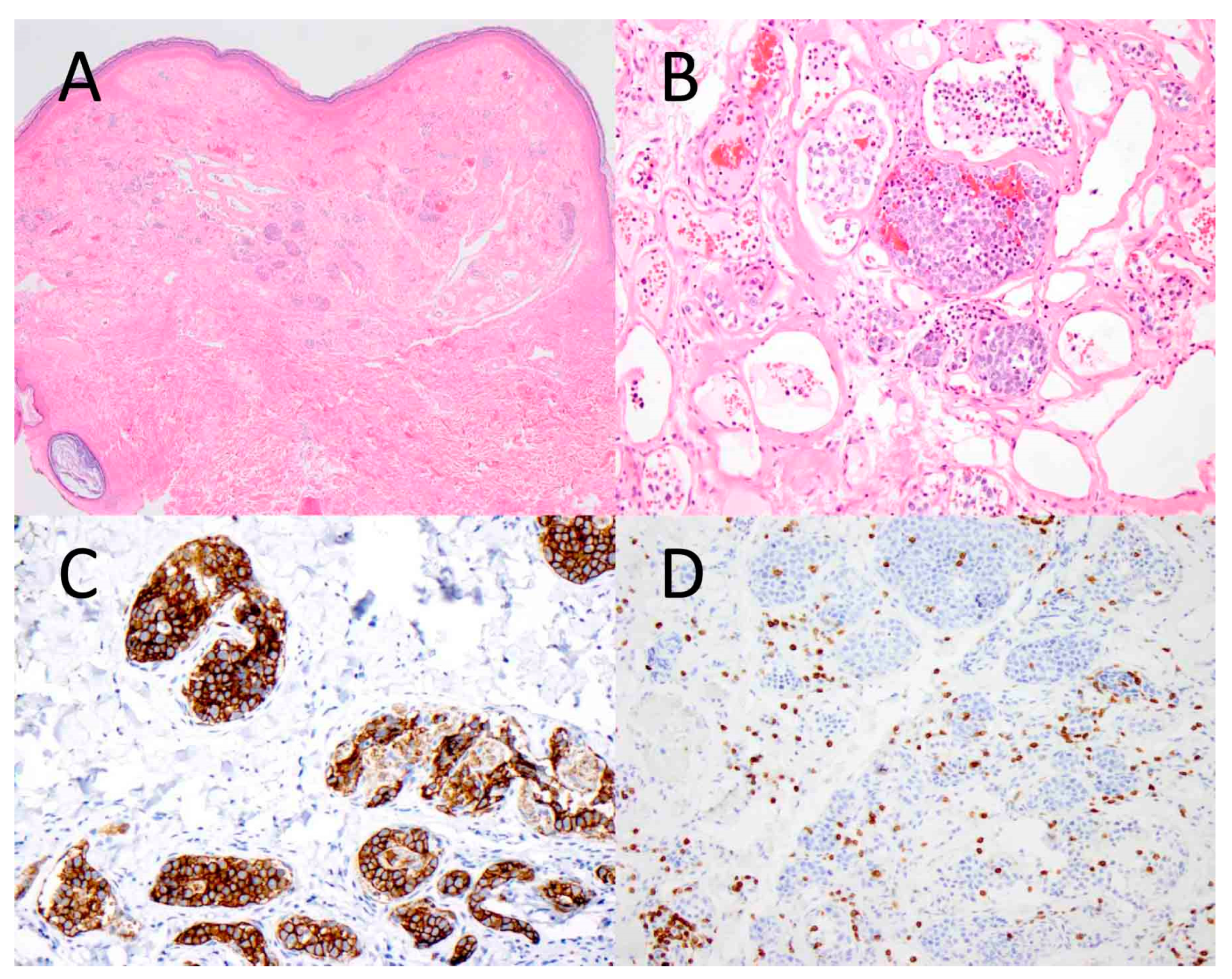
2.2. Histopathology
2.3. Immunophenotype
2.4. Molecular Findings
2.4.1. B-Cell Receptor/Nuclear Factor Kappa Beta Signaling
2.4.2. PDL-1/2
2.5. Pathogenesis
2.6. Summary
3. Intravascular T/NT Lymphoma
3.1. Clinical Presentation
3.1.1. Diagnosis
3.1.2. Treatment and Prognosis
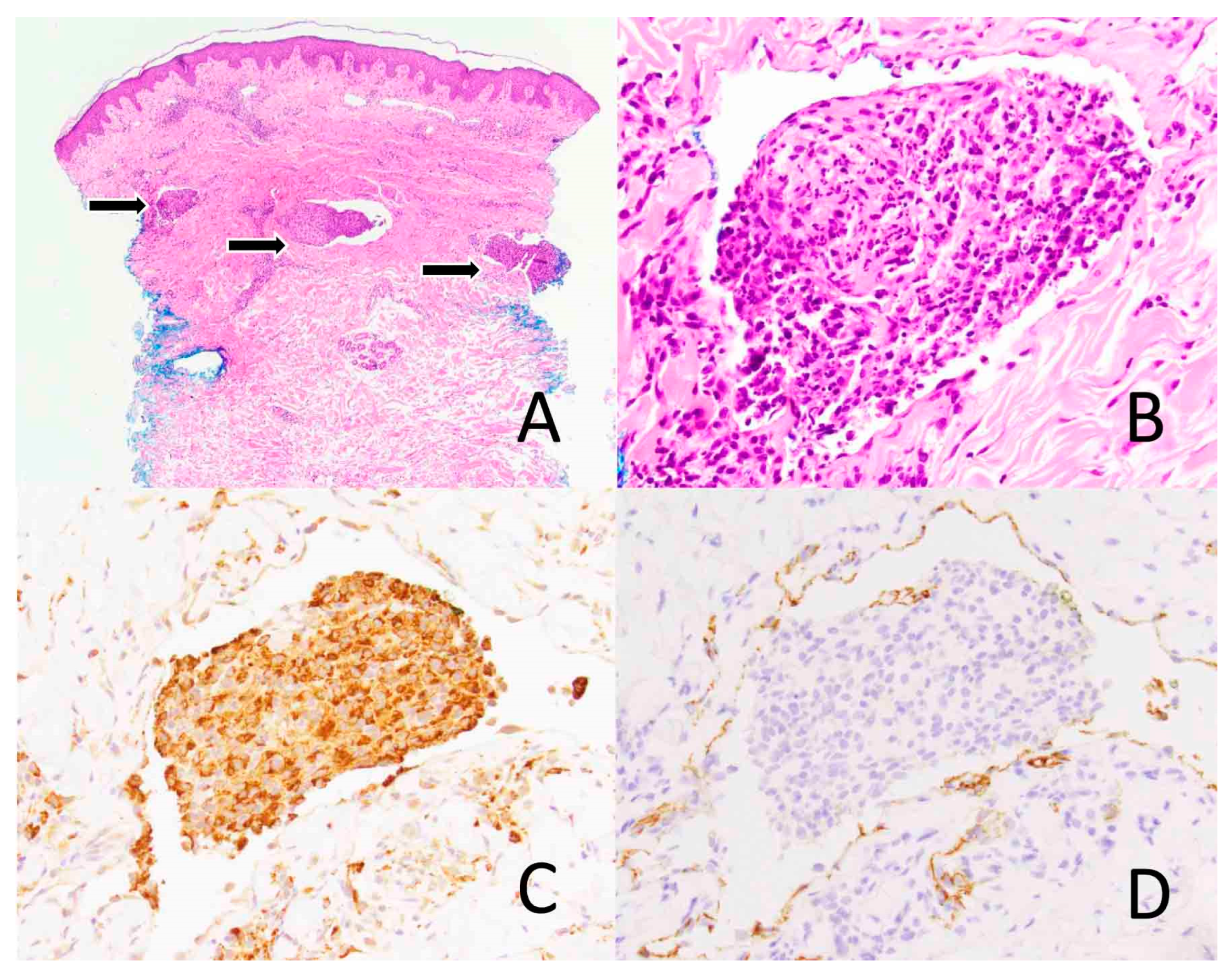
3.2. Histopathology
3.3. Immunophenotype
3.4. Differential Diagnosis
3.5. Molecular Findings
3.6. Pathogenesis
3.7. Summary
4. Intralymphatic Histiocytosis
4.1. Clinical Presentation
4.2. Treatment
4.3. Histopathology
4.4. Immunophenotype
4.5. Differential Diagnosis
4.6. Molecular Findings
4.7. Pathogenesis
4.8. Summary
5. Benign Atypical Intravascular CD30+ T-Cell Proliferation
5.1. Clinical Presentation
5.2. Treatment
5.3. Histopathology
5.4. Differential Diagnosis
5.5. Molecular Findings
5.6. Pathogenesis
5.7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davis, J.W.; Auerbach, A.; Crothers, B.A.; Lewin, E.; Lynch, D.T.; Teschan, N.J.; Schmieg, J.J. Intravascular Large B-Cell Lymphoma. Arch. Pathol. Lab. Med. 2022, 146, 1160–1167. [Google Scholar] [CrossRef]
- Breakell, T.; Waibel, H.; Schliep, S.; Ferstl, B.; Erdmann, M.; Berking, C.; Heppt, M.V. Intravascular Large B-Cell Lymphoma: A Review with a Focus on the Prognostic Value of Skin Involvement. Curr. Oncol. 2022, 29, 2909–2919. [Google Scholar] [CrossRef]
- Rajyaguru, D.J.; Bhaskar, C.; Borgert, A.J.; Smith, A.; Parsons, B. Intravascular Large B-Cell Lymphoma in the United States (US): A Population-Based Study Using Surveillance, Epidemiology, and End Results Program and National Cancer Database. Leuk Lymphoma 2017, 58, 2080–2088. [Google Scholar] [CrossRef]
- Rozenbaum, D.; Tung, J.; Xue, Y.; Hoang, M.P.; Kroshinsky, D. Skin Biopsy in the Diagnosis of Intravascular Lymphoma: A Retrospective Diagnostic Accuracy Study. J. Am. Acad. Dermatol. 2021, 85, 665–670. [Google Scholar] [CrossRef]
- Ferreri, A.J.M.; Dognini, G.P.; Campo, E.; Willemze, R.; Seymour, J.F.; Bairey, O.; Martelli, M.; De Renz, A.O.; Doglioni, C.; Montalbán, C.; et al. Variations in Clinical Presentation, Frequency of Hemophagocytosis and Clinical Behavior of Intravascular Lymphoma Diagnosed in Different Geographical Regions. Haematologica 2007, 92, 486–492. [Google Scholar] [CrossRef]
- Ponzoni, M.; Campo, E.; Nakamura, S. Intravascular Large B-Cell Lymphoma: A Chameleon with Multiple Faces and Many Masks. Blood 2018, 132, 1561–1567. [Google Scholar] [CrossRef]
- Ponzoni, M.; Ferreri, A.J.M.; Campo, E.; Facchetti, F.; Mazzucchelli, L.; Yoshino, T.; Murase, T.; Pileri, S.A.; Doglioni, C.; Zucca, E.; et al. Definition, Diagnosis, and Management of Intravascular Large B-Cell Lymphoma: Proposals and Perspectives from an International Consensus Meeting. J. Clin. Oncol. 2007, 25, 3168–3173. [Google Scholar] [CrossRef]
- Shimada, K.; Yamaguchi, M.; Atsuta, Y.; Matsue, K.; Sato, K.; Kusumoto, S.; Nagai, H.; Takizawa, J.; Fukuhara, N.; Nagafuji, K.; et al. Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, and Prednisolone Combined with High-Dose Methotrexate plus Intrathecal Chemotherapy for Newly Diagnosed Intravascular Large B-Cell Lymphoma (PRIMEUR-IVL): A Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol. 2020, 21, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Farre, B.; Ramis-Zaldivar, J.E.; Castrejón de Anta, N.; Rivas-Delgado, A.; Nadeu, F.; Salmeron-Villalobos, J.; Enjuanes, A.; Karube, K.; Balagué, O.; Cobo, F.; et al. Intravascular Large B-Cell Lymphoma Genomic Profile Is Characterized by Alterations in Genes Regulating NF-κB and Immune Checkpoints. Am. J. Surg. Pathol. 2023, 47, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, A.J.M.; Campo, E.; Seymour, J.F.; Willemze, R.; Ilariucci, F.; Ambrosetti, A.; Zucca, E.; Rossi, G.; López-Guillermo, A.; Pavlovsky, M.A.; et al. Intravascular Lymphoma: Clinical Presentation, Natural History, Management and Prognostic Factors in a Series of 38 Cases, with Special Emphasis on the “Cutaneous Variant”. Br. J. Haematol. 2004, 127, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Geer, M.; Roberts, E.; Shango, M.; Till, B.G.; Smith, S.D.; Abbas, H.; Hill, B.T.; Kaplan, J.; Barr, P.M.; Caimi, P.; et al. Multicentre Retrospective Study of Intravascular Large B-Cell Lymphoma Treated at Academic Institutions within the United States. Br. J. Haematol. 2019, 186, 255–262. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Zhu, Y.; Zhang, W. Prognosis of Intravascular Large B Cell Lymphoma (IVLBCL): Analysis of 182 Patients from Global Case Series. Cancer Manag. Res. 2020, 12, 10531–10540. [Google Scholar] [CrossRef] [PubMed]
- Deyrup, A.; Graves, J.L. Racial Biology and Medical Misconceptions. N. Engl. J. Med. 2022, 386, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, Q.; Wang, D.; Peng, L.; Huang, T.; Ou, C.; Yang, K.; Wang, J. Case Report: Intravascular Large B-Cell Lymphoma: A Clinicopathologic Study of Four Cases With Review of Additional 331 Cases in the Literature. Front. Oncol. 2022, 12, 883141. [Google Scholar] [CrossRef] [PubMed]
- Matsue, K.; Abe, Y.; Kitadate, A.; Miura, D.; Narita, K.; Kobayashi, H.; Takeuchi, M.; Enzan, N.; Tanaka, A.; Takeuchi, K. Sensitivity and Specificity of Incisional Random Skin Biopsy for Diagnosis of Intravascular Large B-Cell Lymphoma. Blood 2019, 133, 1257–1259. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.M.R.; Jansen, P.M.; Willemze, R.; Vermeer, M.H.; Cleton-Jansen, A.-M.; Somers, S.F.; Veelken, H.; van Eijk, R.; Kraan, W.; Kersten, M.J.; et al. High Prevalence of MYD88 and CD79B Mutations in Intravascular Large B-Cell Lymphoma. Blood 2018, 131, 2086–2089. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Yamaguchi, M.; Suzuki, R.; Okamoto, M.; Sato, Y.; Tamaru, J.; Kojima, M.; Miura, I.; Mori, N.; Yoshino, T.; et al. Intravascular Large B-Cell Lymphoma (IVLBCL): A Clinicopathologic Study of 96 Cases with Special Reference to the Immunophenotypic Heterogeneity of CD5. Blood 2007, 109, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Totoraitis, K.; Toama, W.; He, F.; Bohjanen, K.; Williams, S.; Miller, D.D. Concurrent Mycosis Fungoides and Intravascular Large B-Cell Lymphoma in a Single Patient. J. Cutan. Pathol. 2020, 47, 643–648. [Google Scholar] [CrossRef]
- Cenk, H.; Sarac, G.; Karadağ, N.; Berktas, H.B.; Sahin, I.; Sener, S.; Kisaciik, D.; Kapicioglu, Y. Intravascular Lymphoma Presenting with Paraneoplastic Syndrome. Dermatol. Online J. 2020, 26, 13030/qt08252906. [Google Scholar] [CrossRef]
- Ponzoni, M.; Arrigoni, G.; Gould, V.E.; Del Curto, B.; Maggioni, M.; Scapinello, A.; Paolino, S.; Cassisa, A.; Patriarca, C. Lack of CD 29 (Beta1 Integrin) and CD 54 (ICAM-1) Adhesion Molecules in Intravascular Lymphomatosis. Hum. Pathol. 2000, 31, 220–226. [Google Scholar] [CrossRef]
- Kiriakopoulos, A.; Linos, D. Intravascular B-Large Cell Lymphoma: An Unexpected Diagnosis of an Incidental Adrenal Mass. J. Surg. Case Rep. 2019, 2019, rjz048. [Google Scholar] [CrossRef]
- Kusunoki, R.; Fujishiro, H.; Yoshimura, M.; Sawada, K.; Suemitsu, S.; Kataoka, M.; Fujiwara, A.; Tsukano, K.; Kotani, S.; Yamanouchi, S.; et al. Intravascular Large B-Cell Lymphoma Mimicking Hepatobiliary Infection: A Case Report and Literature Review. Intern. Med. 2019, 58, 1885–1889. [Google Scholar] [CrossRef]
- Lim, C.H.; Yoon, S.E.; Kim, W.S.; Lee, K.-H.; Kim, S.J. Imaging Features and Prognostic Value of FDG PET/CT in Patients with Intravascular Large B-Cell Lymphoma. Cancer Manag. Res. 2021, 13, 7289–7297. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, Y.; Zhu, W.; Huo, L.; Zhou, D.; Wang, W.; Wei, C.; Zhang, W. Distinct FDG PET/CT Avidity among Newly Diagnosed Intravascular Large B-Cell Lymphoma Patients: A Descriptive Observational Study. Ann. Hematol. 2024, 103, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.G.; Sheu, S.L.; Kuo, K.Y.; Ally, M.S.; Bailey, E.E.; Kim, J.; Kwong, B.Y. Limited Role of Random Skin Biopsy in the Diagnosis of Intravascular Lymphoma in Adult Patients with Hemophagocytic Lymphohistiocytosis. Acta Haematol. 2017, 138, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Wakida, K.; Takahashi, T.; Ishida, K.; Iwata, H.; Nishida, H. Usefulness of Senile Hemangioma Biopsy for Diagnosis of Intravascular Large B-Cell Lymphoma: A Report of Two Cases and a Literature Review. J. Neurol. Sci. 2017, 373, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Sumi-Mizuno, M.; Fukunaga, A.; Kosaka, H.; Imai, Y.; Nagano, T. Appropriate Indication and Procedure for Random Skin Biopsy in the Diagnosis of Intravascular Large B-Cell Lymphoma. Australas. J. Dermatol. 2021, 62, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Xu, L.; Liu, X.; Wang, Y. Random Skin Biopsy for Diagnosis of Intravascular Large B-Cell Lymphoma: A Case Report and Literature Review. Am. J. Dermatopathol. 2023, 45, 320–322. [Google Scholar] [CrossRef]
- Suehara, Y.; Sakata-Yanagimoto, M.; Hattori, K.; Nanmoku, T.; Itoh, T.; Kaji, D.; Yamamoto, G.; Abe, Y.; Narita, K.; Takeuchi, M.; et al. Liquid Biopsy for the Identification of Intravascular Large B-Cell Lymphoma. Haematologica 2018, 103, e241–e244. [Google Scholar] [CrossRef]
- Shimada, K.; Yoshida, K.; Suzuki, Y.; Iriyama, C.; Inoue, Y.; Sanada, M.; Kataoka, K.; Yuge, M.; Takagi, Y.; Kusumoto, S.; et al. Frequent Genetic Alterations in Immune Checkpoint-Related Genes in Intravascular Large B-Cell Lymphoma. Blood 2021, 137, 1491–1502. [Google Scholar] [CrossRef]
- de Jong, D.; Takeuchi, K.; Ferry, J.A.; Soffietti, R.; Batchelor, T.; Deckert, M.; Hoang-Xuan, K.; Nagane, M.; Shimada, K. Haematolymphoid Tumours; Siebert, R., Dave, S.S., Ott, G., Eds.; WHO Classification of Tumours; International Agency for Research on Cancer (IARC): Lyon, France.
- Liu, Y.; Ma, Y.; Zhou, H.; Zhou, X.; Shao, J. Analysis of Clinicopathological Features and Prognostic Factors of Non-Hodgkin’s Intravascular Large B-Cell Lymphoma. Oncol. Lett. 2020, 20, 43. [Google Scholar] [CrossRef]
- Seegobin, K.; Li, Z.; Alhaj Moustafa, M.; Majeed, U.; Wang, J.; Jiang, L.; Kuhlman, J.; Menke, D.; Li, K.; Kharfan-Dabaja, M.A.; et al. Clinical Characteristics, Prognostic Indicators, and Survival Outcomes in Intravascular Lymphoma: Mayo Clinic Experience (2003–2018). Am. J. Hematol. 2022, 97, 1150–1158. [Google Scholar] [CrossRef]
- Gupta, G.K.; Jaffe, E.S.; Pittaluga, S. A Study of PD-L1 Expression in Intravascular Large B Cell Lymphoma: Correlation with Clinical and Pathological Features. Histopathology 2019, 75, 282–286. [Google Scholar] [CrossRef]
- Qiu, L.; Wang, S.A.; Vega, F.; Khoury, J.D.; Tang, Z.; Garces, S.; Medeiros, L.J.; Thakral, B. From the Archives of MD Anderson Cancer Center: Intravascular Large B-Cell Lymphoma with Numerous Circulating Lymphoma Cells. Ann. Diagn. Pathol. 2022, 58, 151934. [Google Scholar] [CrossRef]
- Patel, N.; Slack, G.W.; Bodo, J.; Ben-Neriah, S.; Villa, D.; Durkin, L.; Socha, D.; Steidl, C.; Hsi, E.D. Immune Escape Mechanisms in Intravascular Large B-Cell Lymphoma: A Molecular Cytogenetic and Immunohistochemical Study. Am. J. Clin. Pathol. 2022, 157, 578–585. [Google Scholar] [CrossRef]
- Bauer, W.M.; Aichelburg, M.C.; Griss, J.; Skrabs, C.; Simonitsch-Klupp, I.; Schiefer, A.I.; Kittler, H.; Jäger, U.; Zeyda, M.; Knobler, R.; et al. Molecular Classification of Tumour Cells in a Patient with Intravascular Large B-Cell Lymphoma. Br. J. Dermatol. 2018, 178, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Shimada, S.; Sugimoto, K.; Nakatochi, M.; Suguro, M.; Hirakawa, A.; Hocking, T.D.; Takeuchi, I.; Tokunaga, T.; Takagi, Y.; et al. Development and Analysis of Patient-Derived Xenograft Mouse Models in Intravascular Large B-Cell Lymphoma. Leukemia 2016, 30, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Kurek, C.; Ehsman, S.; Brighton, T.; Young, K. Incidental Intravascular Large B-Cell Lymphoma in a Renal Cell Carcinoma. Pathology 2020, 52, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, M.; Parente, P.; Sanguedolce, F.; Zizzo, M.; Palicelli, A.; Bisagni, A.; Carosi, I.; Trombetta, D.; Mastracci, L.; Ricci, L.; et al. Intravascular NK/T-Cell Lymphoma: What We Know about This Diagnostically Challenging, Aggressive Disease. Cancers 2022, 14, 5458. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Pimpinelli, N.; Massi, D.; Kadin, M.E.; Meijer, C.J.L.M.; Müller-Hermelink, H.K.; Paulli, M.; Wechsler, J.; Willemze, R.; Audring, H.; et al. Cytotoxic/Natural Killer Cell Cutaneous Lymphomas. Report of EORTC Cutaneous Lymphoma Task Force Workshop. Cancer 2003, 97, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Na, J.M.; Jung, W.; Kim, M.; Cheon, Y.-H.; Lee, J.S.; Song, D.H.; Yang, J.W. Intravascular NK/T-Cell Lymphoma: A Case Report and Literature Review. J. Pathol. Transl. Med. 2023, 57, 332–336. [Google Scholar] [CrossRef]
- Falini, B.; Lazzi, S.; Pileri, S. A Comparison of the International Consensus and 5th WHO Classifications of T-Cell Lymphomas and Histiocytic/Dendritic Cell Tumours. Br. J. Haematol. 2023, 203, 369–383. [Google Scholar] [CrossRef]
- Piccaluga, P.P.; Khattab, S.S. A Comparison of the Fifth World Health Organization and the International Consensus Classifications of Mature T-Cell Lymphomas. Int. J. Mol. Sci. 2023, 24, 14170. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, W.; An, J.; Li, H.; Liu, S. Cutaneous Intravascular Natural Killer-Cell Lymphoma: A Case Report and Review of the Literature. Am. J. Clin. Pathol. 2014, 142, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, F.; Luo, D.; Yao, S.; Chen, Y.; Xu, F.; Luo, X.; He, J.; Liu, Y. Intravascular NK/T-Cell Lymphoma: A Series of Four Cases. Int. J. Clin. Exp. Pathol. 2017, 10, 9541–9550. [Google Scholar]
- Zanelli, M.; Mengoli, M.C.; Del Sordo, R.; Cagini, A.; De Marco, L.; Simonetti, E.; Martino, G.; Zizzo, M.; Ascani, S. Intravascular NK/T-Cell Lymphoma, Epstein-Barr Virus Positive with Multiorgan Involvement: A Clinical Dilemma. BMC Cancer 2018, 18, 1115. [Google Scholar] [CrossRef]
- Wang, J.; Yang, X.; Song, Z.; You, Y. Cutaneous Intravascular NK/T-Cell Lymphoma. Australas. J. Dermatol. 2020, 61, 61–63. [Google Scholar] [CrossRef]
- Vilas Boas, P.; Cerroni, L.; Requena, L. Intravascular Cutaneous Disorders. A Clinicopathologic Review. Am. J. Dermatopathol. 2021, 43, 119–136. [Google Scholar] [CrossRef]
- Amano, S.; Ohta, R.; Sano, C. Natural Killer T Cell Intravascular Lymphoma With Presentation of Musculoskeletal Pain: A Case Report. Cureus 2021, 13, e20711. [Google Scholar] [CrossRef]
- Alhumidi, A. Cutaneous Intravascular NK/T-Cell Lymphoma Mimic Panniculitis Clinically, Case Report and Literature Brief Review. Diagn. Pathol. 2015, 10, 107. [Google Scholar] [CrossRef]
- Vega, F.; Amador, C.; Chadburn, A.; Feldman, A.L.; Hsi, E.D.; Wang, W.; Medeiros, L.J. American Registry of Pathology Expert Opinions: Recommendations for the Diagnostic Workup of Mature T Cell Neoplasms. Ann. Diagn. Pathol. 2020, 49, 151623. [Google Scholar] [CrossRef]
- Okonkwo, L.; Jaffe, E.S. Intravascular Large Cell Lymphoma of NK/T-Cell Type, EBV Positive. Blood 2017, 130, 837. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.; Inamdar, K. Classification and Challenges in the Histopathological Diagnosis of Peripheral T-Cell Lymphomas, Emphasis on the WHO-HAEM5 Updates. Front. Oncol. 2022, 12, 1099265. [Google Scholar] [CrossRef]
- Bi, Y.; Huo, Z.; Liang, Z.; Meng, Y.; Jia, C.; Shi, X.; Song, L.; Luo, Y.; Ling, Q.; Liu, T. Intravascular NK-Cell Lymphoma: A Case Report and Review of the Literature. Diagn. Pathol. 2015, 10, 84. [Google Scholar] [CrossRef]
- Nakamichi, N.; Fukuhara, S.; Aozasa, K.; Morii, E. NK-Cell Intravascular Lymphomatosis--a Mini-Review. Eur. J. Haematol. 2008, 81, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.P.; Ahearne, M.J.; Pettengell, R.; Dearden, C.; El-Sharkawi, D.; Kassam, S.; Cook, L.; Cwynarski, K.; Illidge, T.; Collins, G. Guidelines for the Management of Mature T- and Natural Killer-Cell Lymphomas (Excluding Cutaneous T-Cell Lymphoma): A British Society for Haematology Guideline. Br. J. Haematol. 2022, 196, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.; Schmitt, M.; Andrulis, M.; Schweizer, L.; Dietrich, S.; Alber, B.; Harting, I.; Kurz, F.T.; Martens, U.M.; Ho, A.D.; et al. Cure of Intravascular NK/T-Cell Lymphoma of the Central Nervous System by Allogeneic Hematopoietic Cell Transplantation. Bone Marrow Transpl. 2022, 57, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, K.; Yamashita, D.; Sakamoto, R.; Ishikawa, T.; Chuang, S.-S.; Itoh, T.; Imai, Y. Intravascular NK/T-Cell Lymphoma: Clinicopathological and Integrated Molecular Analysis of Two Cases Provides a Clue to Disease Pathogenesis. J. Clin. Pathol. 2019, 72, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Cerroni, L.; Massone, C.; Kutzner, H.; Mentzel, T.; Umbert, P.; Kerl, H. Intravascular Large T-Cell or NK-Cell Lymphoma: A Rare Variant of Intravascular Large Cell Lymphoma with Frequent Cytotoxic Phenotype and Association with Epstein-Barr Virus Infection. Am. J. Surg. Pathol. 2008, 32, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Alegría-Landa, V.; Manzarbeitia, F.; Salvatierra Calderón, M.G.; Requena, L.; Rodríguez-Pinilla, S.M. Cutaneous Intravascular Natural Killer/T Cell Lymphoma with Peculiar Immunophenotype. Histopathology 2017, 71, 994–1002. [Google Scholar] [CrossRef]
- Melchers, R.C.; Willemze, R.; Jansen, P.M.; Daniëls, L.A.; Vermeer, M.H.; Quint, K.D. A Rare Case of Cutaneous Epstein-Barr Virus-Negative Intravascular Cytotoxic T-Cell Lymphoma. JAAD Case Rep. 2019, 5, 548–551. [Google Scholar] [CrossRef]
- Syrykh, C.; Péricart, S.; Lamaison, C.; Escudié, F.; Brousset, P.; Laurent, C. Epstein-Barr Virus-Associated T- and NK-Cell Lymphoproliferative Diseases: A Review of Clinical and Pathological Features. Cancers 2021, 13, 3315. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Romero, C.; Bologna-Molina, R.; Paes de Almeida, O.; Santos-Silva, A.R.; Prado-Ribeiro, A.C.; Brandão, T.B.; Carlos, R. Extranodal NK/T Cell Lymphoma, Nasal Type: An Updated Overview. Crit. Rev. Oncol. Hematol. 2021, 159, 103237. [Google Scholar] [CrossRef] [PubMed]
- Au, W.; Weisenburger, D.D.; Intragumtornchai, T.; Nakamura, S.; Kim, W.-S.; Sng, I.; Vose, J.; Armitage, J.O.; Liang, R. International Peripheral T-Cell Lymphoma Project Clinical Differences between Nasal and Extranasal Natural Killer/T-Cell Lymphoma: A Study of 136 Cases from the International Peripheral T-Cell Lymphoma Project. Blood 2009, 113, 3931–3937. [Google Scholar] [CrossRef]
- Kim, W.Y.; Montes-Mojarro, I.A.; Fend, F.; Quintanilla-Martinez, L. Epstein-Barr Virus-Associated T and NK-Cell Lymphoproliferative Diseases. Front. Pediatr. 2019, 7, 71. [Google Scholar] [CrossRef]
- Wu, X.; Li, P.; Zhao, J.; Yang, X.; Wang, F.; Yang, Y.Q.; Fang, F.; Xu, Y.; Zhang, H.; Wang, W.Y.; et al. A Clinical Study of 115 Patients with Extranodal Natural Killer/T-Cell Lymphoma, Nasal Type. Clin. Oncol. (R. Coll. Radiol.) 2008, 20, 619–625. [Google Scholar] [CrossRef]
- Suzuki, R. NK/T Cell Lymphoma: Updates in Therapy. Curr. Hematol. Malig. Rep. 2018, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Amoh, Y. Case of Extranodal Natural Killer/T-Cell Lymphoma, Nasal Type, Presenting With Intravascular Localization of Tumor Cells in Skin Biopsies From Both Plaque and Normal-Appearing Skin: A Case Report and Literature Review. Am. J. Dermatopathol. 2020, 42, 196–203. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Swerdlow, S.H.; Tousseyn, T.; Barrionuevo, C.; Nakamura, S.; Jaffe, E.S. New Concepts in EBV-Associated B, T, and NK Cell Lymphoproliferative Disorders. Virchows Arch. 2023, 482, 227–244. [Google Scholar] [CrossRef]
- Samols, M.A.; Su, A.; Ra, S.; Cappel, M.A.; Louissant, A.; Knudson, R.A.; Ketterling, R.P.; Said, J.; Binder, S.; Harris, N.L.; et al. Intralymphatic Cutaneous Anaplastic Large Cell Lymphoma/Lymphomatoid Papulosis: Expanding the Spectrum of CD30-Positive Lymphoproliferative Disorders. Am. J. Surg. Pathol. 2014, 38, 1203–1211. [Google Scholar] [CrossRef]
- Fujikura, K.; Yoshida, M.; Uesaka, K. Transcriptome Complexity in Intravascular NK/T-Cell Lymphoma. J. Clin. Pathol. 2020, 73, 671–675. [Google Scholar] [CrossRef]
- Fujikura, K.; Yamashita, D.; Yoshida, M.; Ishikawa, T.; Itoh, T.; Imai, Y. Cytogenetic Complexity and Heterogeneity in Intravascular Lymphoma. J. Clin. Pathol. 2021, 74, 244–250. [Google Scholar] [CrossRef]
- O’Grady, J.T.; Shahidullah, H.; Doherty, V.R.; al-Nafussi, A. Intravascular Histiocytosis. Histopathology 1994, 24, 265–268. [Google Scholar] [CrossRef]
- Bakr, F.; Webber, N.; Fassihi, H.; Swale, V.; Lewis, F.; Rytina, E.; Ben-Zvi, G.T.; Norris, P.; Espinosa, O.; Dhar, S.; et al. Primary and Secondary Intralymphatic Histiocytosis. J. Am. Acad. Dermatol. 2014, 70, 927–933. [Google Scholar] [CrossRef]
- Rieger, E.; Soyer, H.P.; Leboit, P.E.; Metze, D.; Slovak, R.; Kerl, H. Reactive Angioendotheliomatosis or Intravascular Histiocytosis? An Immunohistochemical and Ultrastructural Study in Two Cases of Intravascular Histiocytic Cell Proliferation. Br. J. Dermatol. 1999, 140, 497–504. [Google Scholar] [CrossRef]
- Pruim, B.; Strutton, G.; Congdon, S.; Whitehead, K.; Donaldson, E. Cutaneous Histiocytic Lymphangitis: An Unusual Manifestation of Rheumatoid Arthritis. Australas. J. Dermatol. 2000, 41, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Takiwaki, H.; Adachi, A.; Kohno, H.; Ogawa, Y. Intravascular or Intralymphatic Histiocytosis Associated with Rheumatoid Arthritis: A Report of 4 Cases. J. Am. Acad. Dermatol. 2004, 50, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, A.; Asada, H.; Niizeki, H.; Nonomura, A.; Miyagawa, S. Intravascular Histiocytosis Associated with Rheumatoid Arthritis: Report of a Case with Lymphatic Endothelial Proliferation. Br. J. Dermatol. 2005, 152, 1385–1387. [Google Scholar] [CrossRef] [PubMed]
- Requena, L.; El-Shabrawi-Caelen, L.; Walsh, S.N.; Segura, S.; Ziemer, M.; Hurt, M.A.; Sangüeza, O.P.; Kutzner, H. Intralymphatic Histiocytosis. A Clinicopathologic Study of 16 Cases. Am. J. Dermatopathol. 2009, 31, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Crowson, A.N. The Spectrum of Cutaneous Lesions in Rheumatoid Arthritis: A Clinical and Pathological Study of 43 Patients. J. Cutan. Pathol. 2003, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nishie, W.; Sawamura, D.; Iitoyo, M.; Shimizu, H. Intravascular Histiocytosis Associated with Rheumatoid Arthritis. Dermatology 2008, 217, 144–145. [Google Scholar] [CrossRef]
- Demirkesen, C.; Kran, T.; Leblebici, C.; Yücelten, D.; Aksu, A.E.K.; Mat, C. Intravascular/Intralymphatic Histiocytosis: A Report of 3 Cases. Am. J. Dermatopathol. 2015, 37, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Washio, K.; Nakata, K.; Nakamura, A.; Horikawa, T. Pressure Bandage as an Effective Treatment for Intralymphatic Histiocytosis Associated with Rheumatoid Arthritis. Dermatology 2011, 223, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Kang, H.J.; Kim, M.-S.; Jue, M.-S. Intralymphatic Histiocytosis Associated with Osteoarthritis: A Case Report. Ann. Dermatol. 2022, 34, 225–227. [Google Scholar] [CrossRef]
- Korman, J.B.; Burgin, S.; Tahan, S.R. Intralymphatic Histiocytosis in Association with Severe Osteoarthritis of the Shoulder. J. Am. Acad. Dermatol. 2013, 69, e314–e315. [Google Scholar] [CrossRef]
- Ríos-Viñuela, E.; Bernia, E.; Diago, A.; Traves, V.; Requena, C.; Llombart, B.; Sanmartín, O. Cutaneous Intralymphatic Histiocytosis Associated with Breast and Orthopedic Surgery. J. Cutan. Pathol. 2021, 48, 725–729. [Google Scholar] [CrossRef]
- Chiu, Y.E.; Maloney, J.E.; Bengana, C. Erythematous Patch Overlying a Swollen Knee--Quiz Case. Intralymphatic Histiocytosis. Arch. Dermatol. 2010, 146, 1037–1042. [Google Scholar] [CrossRef]
- Grekin, S.; Mesfin, M.; Kang, S.; Fullen, D.R. Intralymphatic Histiocytosis Following Placement of a Metal Implant. J. Cutan. Pathol. 2011, 38, 351–353. [Google Scholar] [CrossRef]
- Abuawad, Y.G.; Diniz, T.d.A.C.B.; Kakizaki, P.; Valente, N.Y.S. Intravascular Histiocytosis: Case Report of a Rare Disease Probably Associated with Silicone Breast Implant. An. Bras. Dermatol. 2020, 95, 347–350. [Google Scholar] [CrossRef]
- Goldsmith, J.F.; Tahan, S.R. Intralymphatic Histiocytosis in Healing Cellulitis: Case Report and Review of the Literature. J. Cutan. Pathol. 2020, 47, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Pouryazdanparast, P.; Yu, L.; Dalton, V.K.; Haefner, H.K.; Brincat, C.; Mandell, S.H.; Cho, K.R.; Fullen, D.R. Intravascular Histiocytosis Presenting with Extensive Vulvar Necrosis. J. Cutan. Pathol. 2009, 36 (Suppl. S1), 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Nagai, H.; Tsuji, G.; Morinobu, A.; Kumagai, S.; Nishigori, C. Effectiveness of Infliximab for Intralymphatic Histiocytosis with Rheumatoid Arthritis. Arch. Dermatol. 2011, 147, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.J.; Ingersoll, Z.; Blackwell, M. Intralymphatic Histiocytosis: An Unusual Presentation. Case Rep. Dermatol. 2021, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Catalina-Fernández, I.; Alvárez, A.C.; Martin, F.C.; Fernández-Mera, J.J.; Sáenz-Santamaría, J. Cutaneous Intralymphatic Histiocytosis Associated with Rheumatoid Arthritis: Report of a Case and Review of the Literature. Am. J. Dermatopathol. 2007, 29, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Kahn, H.J.; Bailey, D.; Marks, A. Monoclonal Antibody D2-40, a New Marker of Lymphatic Endothelium, Reacts with Kaposi’s Sarcoma and a Subset of Angiosarcomas. Mod. Pathol. 2002, 15, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Mensing, C.H.; Krengel, S.; Tronnier, M.; Wolff, H.H. Reactive Angioendotheliomatosis: Is It “Intravascular Histiocytosis”? J. Eur. Acad. Dermatol. Venereol. 2005, 19, 216–219. [Google Scholar] [CrossRef]
- Mazloom, S.E.; Stallings, A.; Kyei, A. Differentiating Intralymphatic Histiocytosis, Intravascular Histiocytosis, and Subtypes of Reactive Angioendotheliomatosis: Review of Clinical and Histologic Features of All Cases Reported to Date. Am. J. Dermatopathol. 2017, 39, 33–39. [Google Scholar] [CrossRef]
- Okamoto, N.; Tanioka, M.; Yamamoto, T.; Shiomi, T.; Miyachi, Y.; Utani, A. Intralymphatic Histiocytosis Associated with Rheumatoid Arthritis. Clin. Exp. Dermatol. 2008, 33, 516–518. [Google Scholar] [CrossRef]
- Asagoe, K.; Torigoe, R.; Ofuji, R.; Iwatsuki, K. Reactive Intravascular Histiocytosis Associated with Tonsillitis. Br. J. Dermatol. 2006, 154, 560–563. [Google Scholar] [CrossRef]
- Ellis, T.M.; Simms, P.E.; Slivnick, D.J.; Jäck, H.M.; Fisher, R.I. CD30 Is a Signal-Transducing Molecule That Defines a Subset of Human Activated CD45RO+ T Cells. J. Immunol. 1993, 151, 2380–2389. [Google Scholar] [CrossRef]
- Del Prete, G.; De Carli, M.; Almerigogna, F.; Daniel, C.K.; D’Elios, M.M.; Zancuoghi, G.; Vinante, F.; Pizzolo, G.; Romagnani, S. Preferential Expression of CD30 by Human CD4+ T Cells Producing Th2-Type Cytokines. FASEB J. 1995, 9, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Werner, B.; Massone, C.; Kerl, H.; Cerroni, L. Large CD30-Positive Cells in Benign, Atypical Lymphoid Infiltrates of the Skin. J. Cutan Pathol. 2008, 35, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, L.T.; Pieretti, M.; Chapman, S.F.; Horenstein, M.G. CD30-Positive Atypical Lymphoid Cells in Common Non-Neoplastic Cutaneous Infiltrates Rich in Neutrophils and Eosinophils. Am. J. Surg. Pathol. 2003, 27, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.L.; Stone, M.S.; Liu, V. Atypical Intravascular CD30+ T-Cell Proliferation Following Trauma in a Healthy 17-Year-Old Male: First Reported Case of a Potential Diagnostic Pitfall and Literature Review. J. Cutan. Pathol. 2009, 36, 350–354. [Google Scholar] [CrossRef]
- Jang, N.R.; Kim, M.K.; Shin, D.H.; Gu, M.J. Benign Atypical Intralymphatic CD30+ T-Cell Proliferation: A Case Report and Literature Review. Ann. Dermatol. 2019, 31, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Falkenbach, E.; Fernández-Figueras, M.T.; Rodríguez-Peralto, J.L. Benign Atypical Intravascular CD30(+) T-Cell Proliferation: A Reactive Condition Mimicking Intravascular Lymphoma. Am. J. Dermatopathol. 2013, 35, 143–150. [Google Scholar] [CrossRef]
- Kempf, W.; Keller, K.; John, H.; Dommann-Scherrer, C. Benign Atypical Intravascular CD30+ T-Cell Proliferation: A Recently Described Reactive Lymphoproliferative Process and Simulator of Intravascular Lymphoma: Report of a Case Associated with Lichen Sclerosus and Review of the Literature. Am. J. Clin. Pathol. 2014, 142, 694–699. [Google Scholar] [CrossRef]
- Ardighieri, L.; Lonardi, S.; Vermi, W.; Medicina, D.; Cerroni, L.; Facchetti, F. Intralymphatic Atypical T-Cell Proliferation in a Cutaneous Hemangioma. J. Cutan. Pathol. 2010, 37, 497–503. [Google Scholar] [CrossRef]
- Ferrisse, T.M.; Almeida, L.Y.; Silveira, H.A.; de Oliveira, A.B.; Bufalino, A.; León, J.E. Benign Atypical Intralymphatic CD30+ Lymphoid Proliferation with Activated Regulatory T-Cell Phenotype in the Oral Cavity. J. Cutan. Pathol. 2019, 46, 891–894. [Google Scholar] [CrossRef]
- Calamaro, P.; Cerroni, L. Intralymphatic Proliferation of T-Cell Lymphoid Blasts in the Setting of Hidradenitis Suppurativa. Am. J. Dermatopathol. 2016, 38, 536–540. [Google Scholar] [CrossRef]
- Hung, W.-K.; Chen, C.-B.; Kuo, T.-T.; Chen, W.-T. Pembrolizumab-Induced Benign Atypical Intralymphatic CD30+ T-Cell Proliferation Mimicking Intravascular Lymphoma. J. Dermatol. 2022, 49, e53–e54. [Google Scholar] [CrossRef]
- Bush, A.E.; Garcia, A.; Li, J.; Curry, J.; Chon, S.Y. CD30+ Lymphomatoid Skin Toxicity Secondary to Ipilimumab. JAAD Case Rep. 2020, 6, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Weingertner, N.; Mitcov, M.; Chenard, M.-P.; Cribier, B. Intralymphatic CD30+ T-Cell Proliferation during DRESS: A Mimic of Intravascular Lymphoma. J. Cutan. Pathol. 2016, 43, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Takeichi, T.; Nishida, T.; Sato, J.; Takahashi, Y.; Yamamura, M.; Ogi, T.; Akiyama, M. Extensive Multiple Organ Involvement in VEXAS Syndrome. Arthritis Rheumatol. 2021, 73, 1896–1897. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Han, J.H.; Go, J.H.; Kim, D.S.; Kwon, O.J.; Yang, W.I.; Shin, D.H.; Ree, H.J. Intravascular Lymphomatosis: A Clinicopathological Study of Two Cases Presenting as an Interstitial Lung Disease. Histopathology 1997, 31, 555–562. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Clinical features |
|
| Histopathology |
|
| Clinical features |
|
| Histopathology |
|
| Clinical features |
|
| Histopathology |
|
| Clinical features |
|
| Histopathology |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall, E.H.; Brumbaugh, B.; Holt, A.; Chen, S.T.; Hoang, M.P. Cutaneous Intravascular Hematolymphoid Entities: A Review. Diagnostics 2024, 14, 679. https://doi.org/10.3390/diagnostics14070679
Marshall EH, Brumbaugh B, Holt A, Chen ST, Hoang MP. Cutaneous Intravascular Hematolymphoid Entities: A Review. Diagnostics. 2024; 14(7):679. https://doi.org/10.3390/diagnostics14070679
Chicago/Turabian StyleMarshall, Emily Hatheway, Bethany Brumbaugh, Allison Holt, Steven T. Chen, and Mai P. Hoang. 2024. "Cutaneous Intravascular Hematolymphoid Entities: A Review" Diagnostics 14, no. 7: 679. https://doi.org/10.3390/diagnostics14070679
APA StyleMarshall, E. H., Brumbaugh, B., Holt, A., Chen, S. T., & Hoang, M. P. (2024). Cutaneous Intravascular Hematolymphoid Entities: A Review. Diagnostics, 14(7), 679. https://doi.org/10.3390/diagnostics14070679