
Congenital Heart Malformations Masked by Infantile Gangliosidosis—Case Report and Growing Evidence for Metabolic Disease-Associated Aortopathies


 , , , and
, , , and
Abstract
1. Introduction
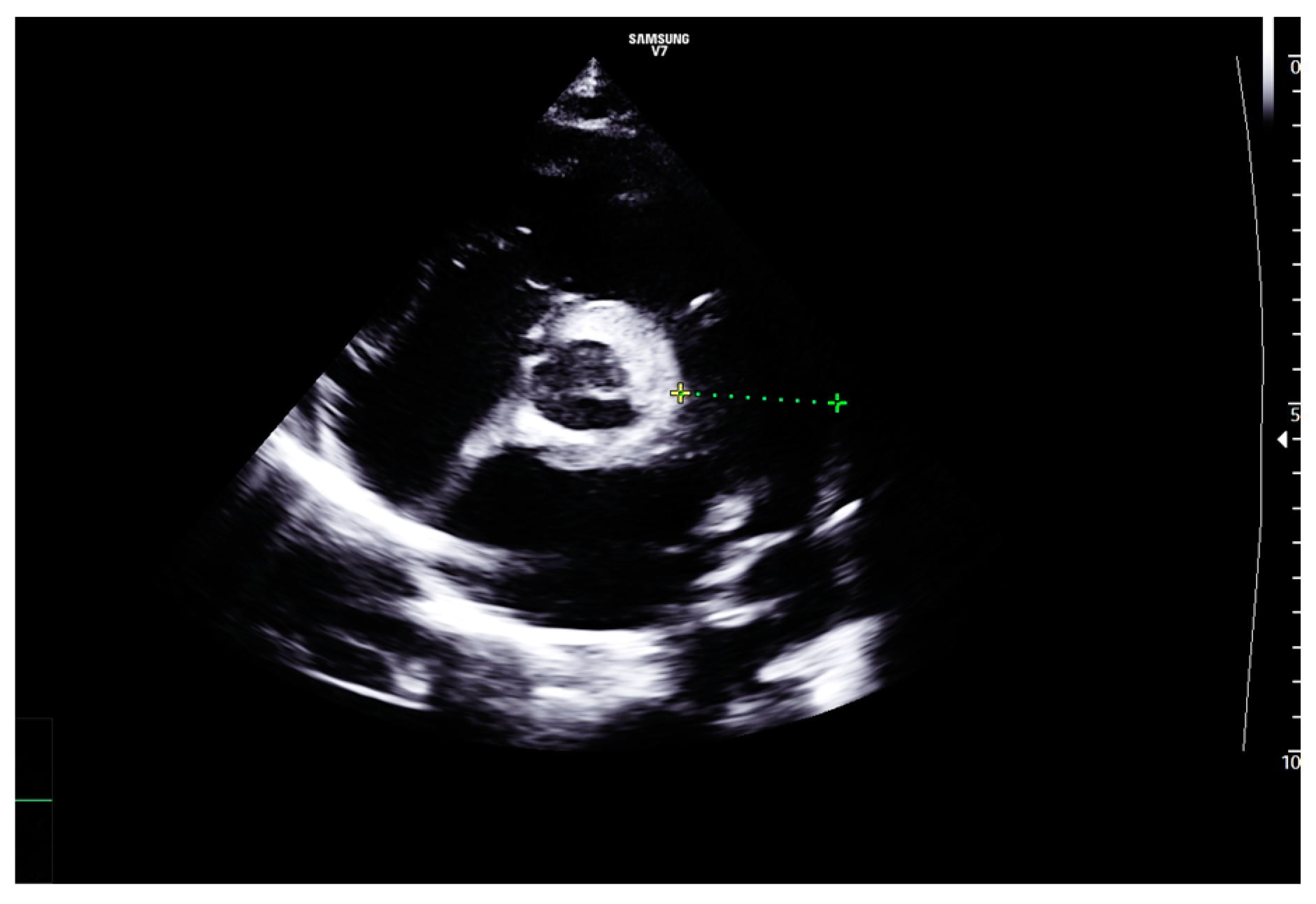
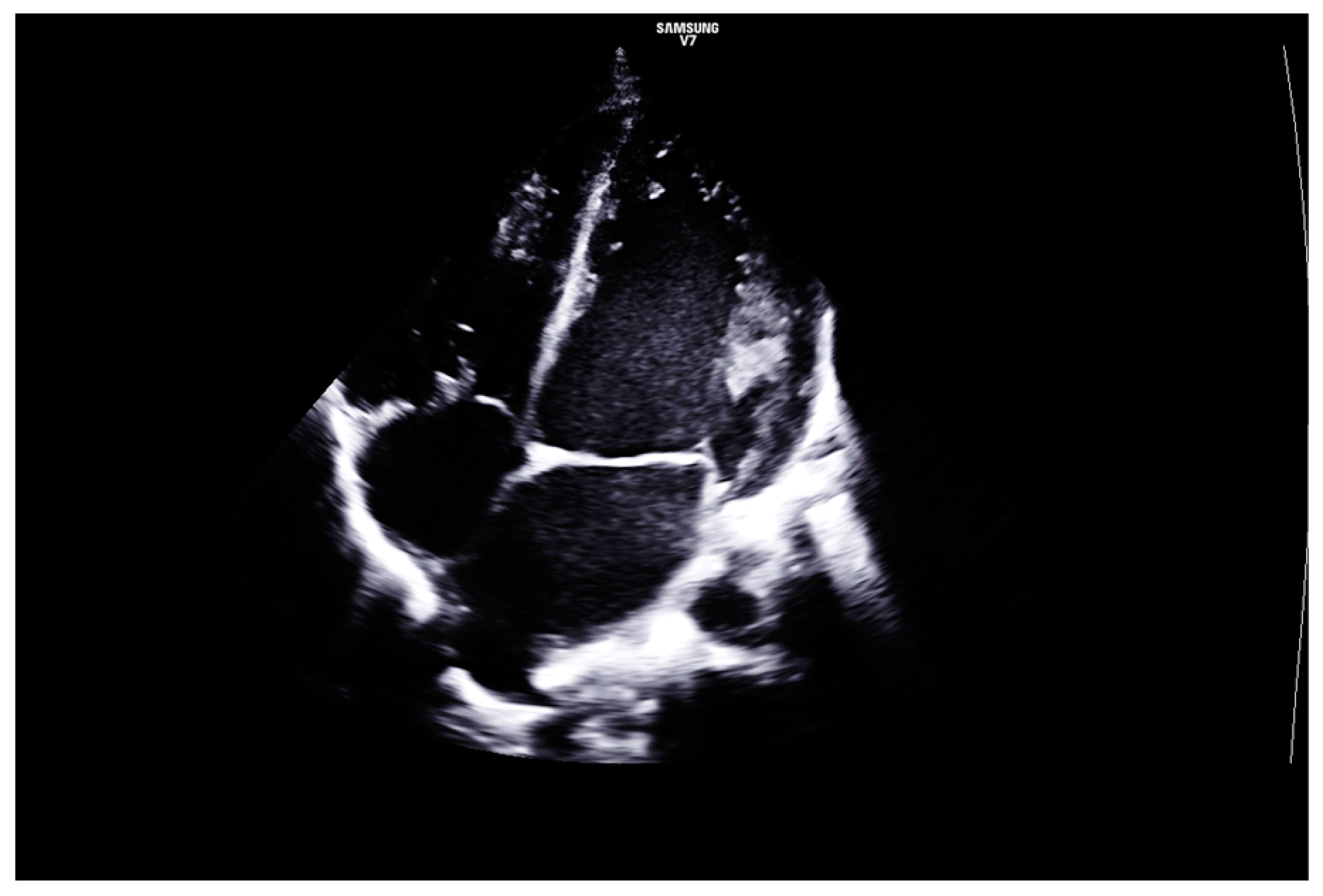
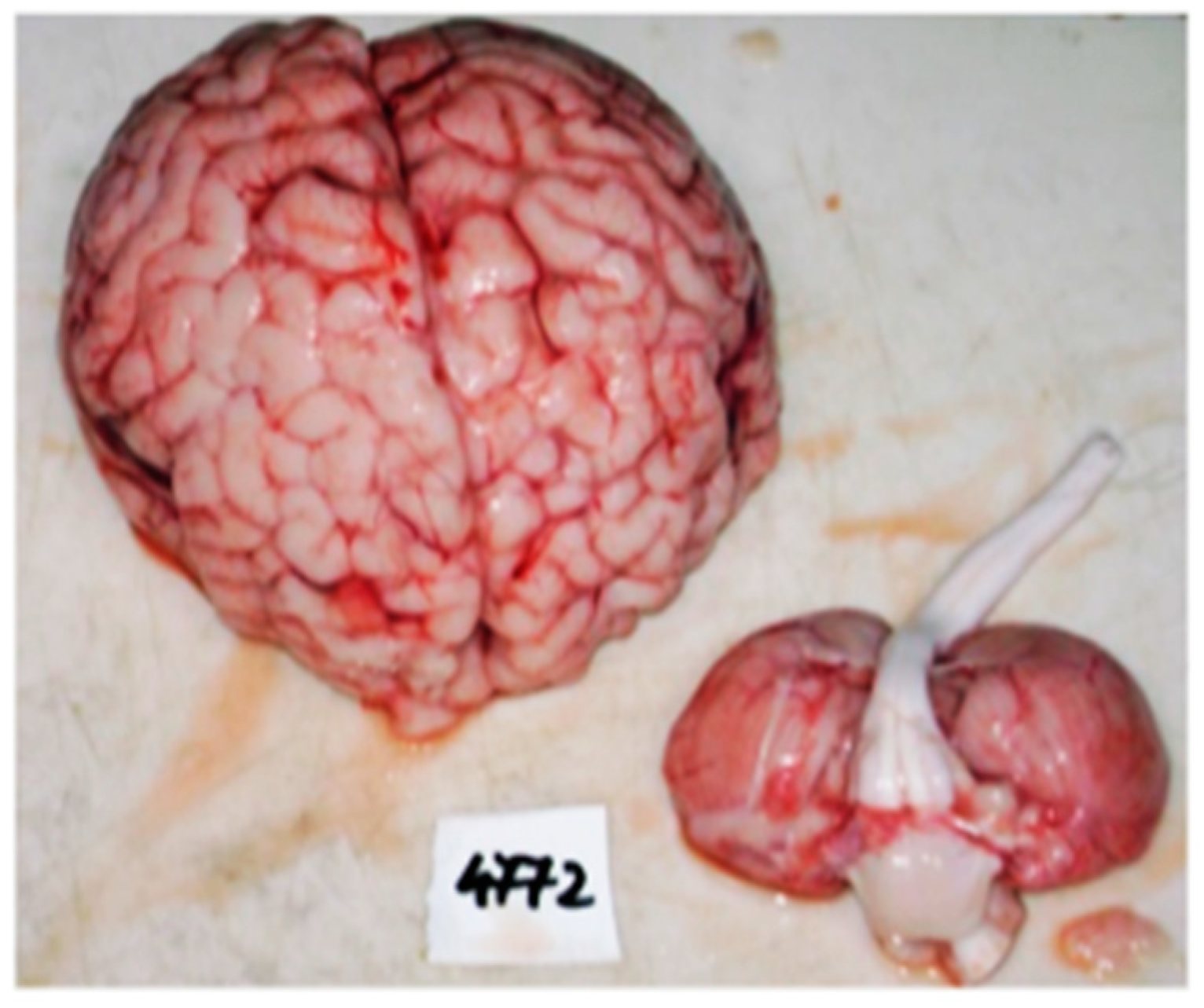
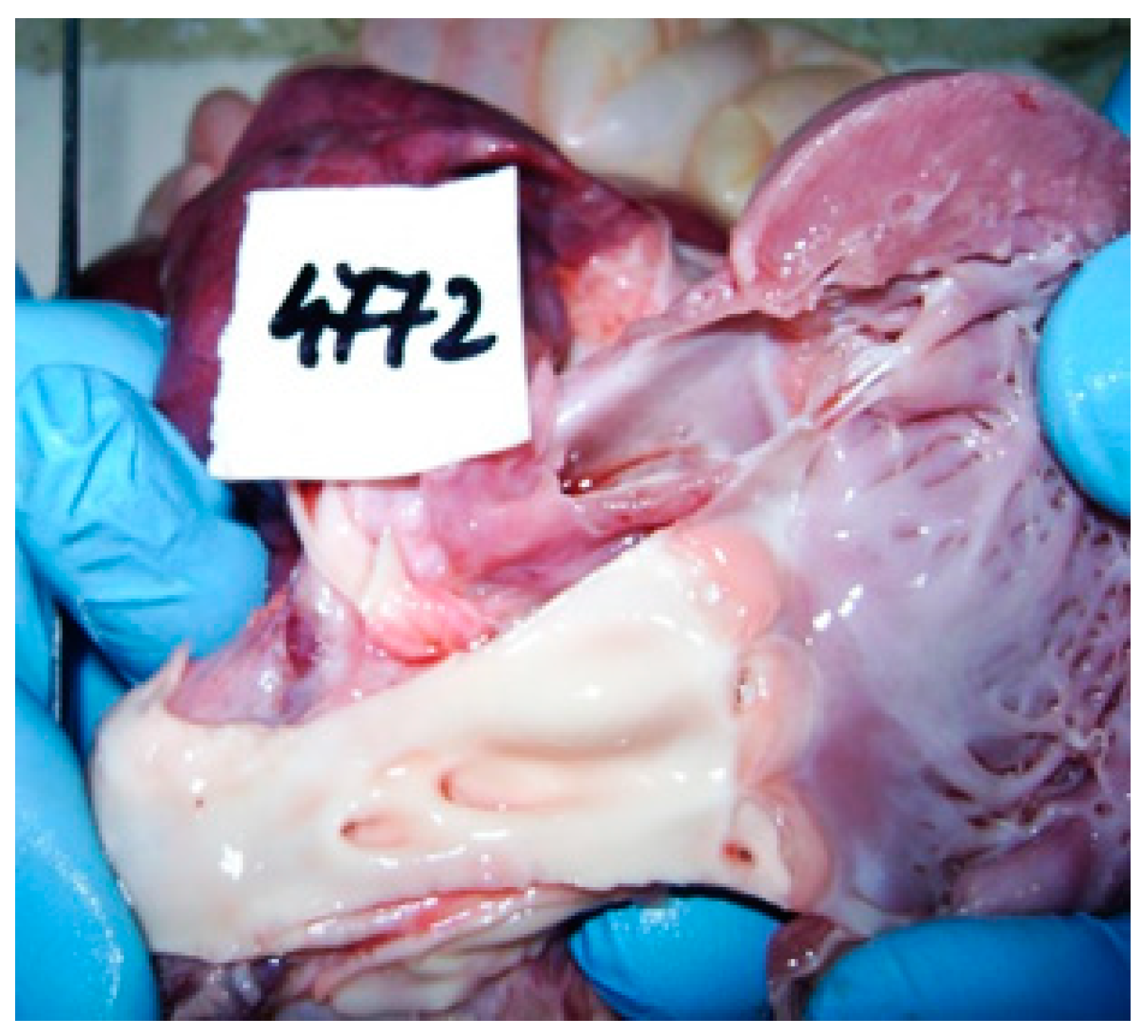
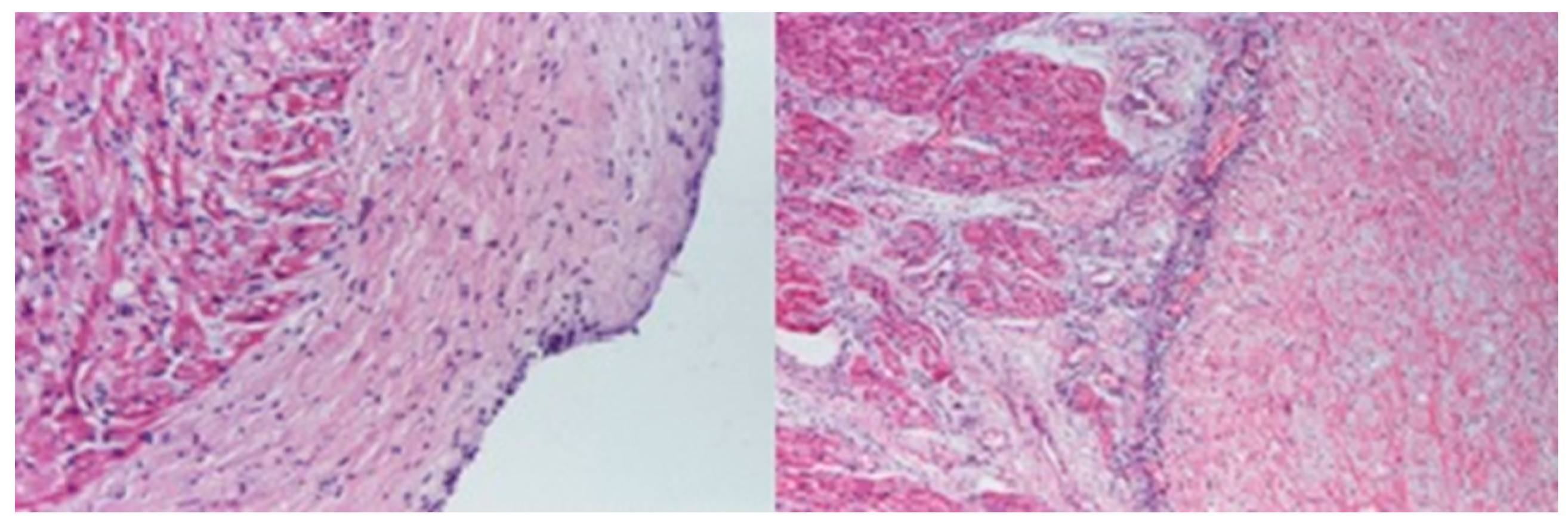
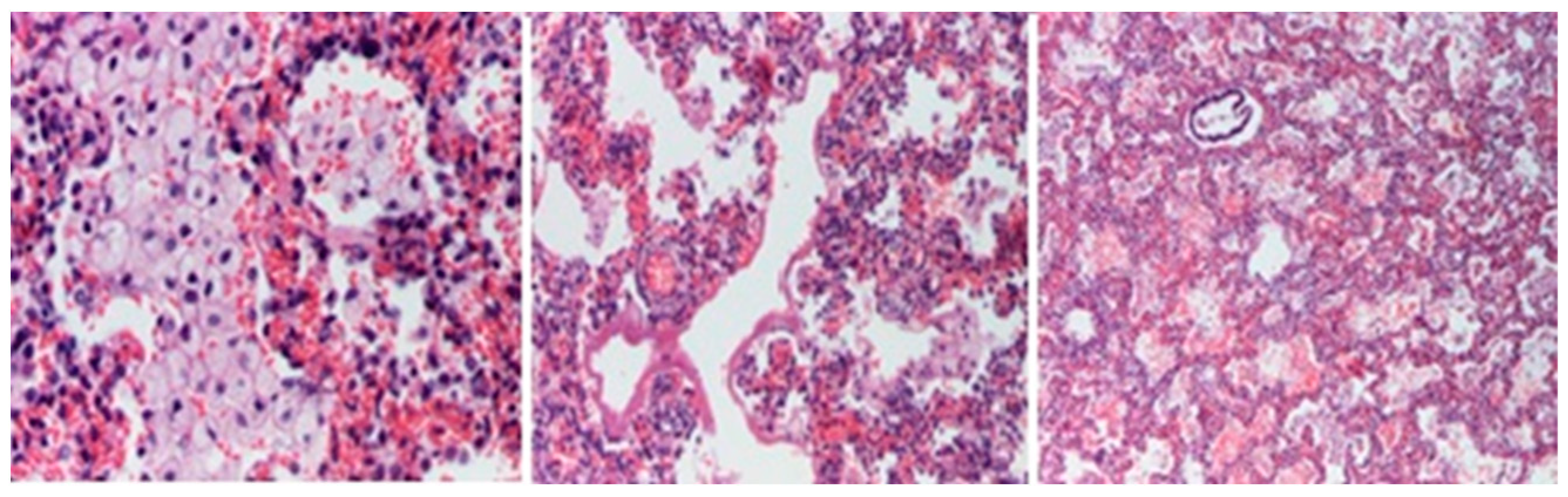
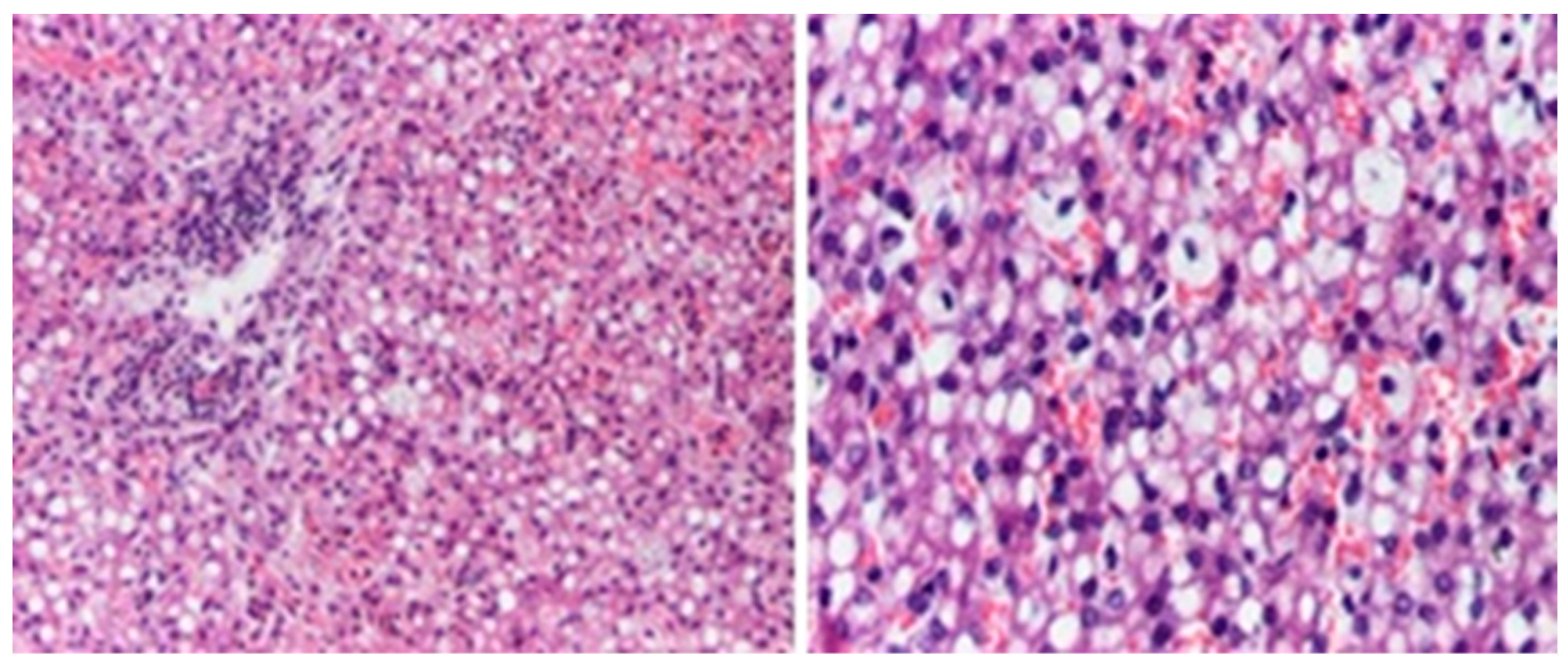
2. Case Report
3. Discussions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blau, N.; Duran, M.; Gibson, M.K.; Dionisi-Vici, C. Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Tanpaiboon, P. Practical Management of Lysosomal Storage Disorders (LSDs). Transl. Sci. Rare 2019, 4, 133–157. [Google Scholar] [CrossRef]
- Kingma, S.D.K.; Ceulemans, B.; Kenis, S.; Jonckheere, A.I. Are GMI gangliosidosis and Morquio type B two different disorders or part of one phenotypic spectrum? JIMD Rep. 2021, 59, 90–103. [Google Scholar] [CrossRef]
- Regier, D.S.; Tifft, C.J.; Rothermel, C.E. GLB1-Related Disorders. In GeneReviews® [Internet]; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemyia, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK164500/ (accessed on 15 January 2024).
- Yuskiv, N.; Higaki, K.; Stockler-Ipsiroglu, S. Morquio B Disease. Disease Characteristics and Treatment Options of a Distinct GLB1-Related Dysostosis Multiplex. Int. J. Mol. Sci. 2020, 21, 9121. [Google Scholar] [CrossRef]
- Front, S.; Biela-Banaś, A.; Burda, P.; Ballhausen, D.; Higaki, K.; Caciotti, A.; Morrone, A.; Charollais-Thoenig, J.; Gallienne, E.; Demotz, S.; et al. (5aR)-5a-C-Pentyl-4-epi-isofagomine: A powerful inhibitor of lysosomal β-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur. J. Med. Chem. 2017, 126, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Stütz, A.E.; Thonhofer, M.; Weber, P.; Wolfsgruber, A.; Wrodnigg, T.M. Pharmacological Chaperones for β-Galactosidase Related to GM1-Gangliosidosis and Morquio B: Recent Advances. Chem. Rec. 2021, 21, 2980. [Google Scholar] [CrossRef]
- Mak, J.; Cowan, T.M. Detecting lysosomal storage disorders by glycomic profiling using liquid chromatography mass spectrometry. Mol. Genet. Metab. 2021, 134, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Nieto, W.G.; Candelo, E.; Pachajoa, H.; Alméciga-Díaz, C.J. Hematological Findings in Lysosomal Storage Disorders: A Perspective from the Medical Laboratory. eJIFCC 2022, 33, 28–42. [Google Scholar]
- Lang, F.M.; Korner, P.; Harnett, M.; Karunakara, A.; Tifft, C.J. The natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis. Mol. Genet. Metab. 2020, 129, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Carubbi, F.; Barbato, A.; Burlina, A.B.; Francini, F.; Mignani, R.; Pegoraro, E.; Landini, L.; De Danieli, G.; Bruni, S.; Strazzullo, P. Italian Society of Human Nutrition Working Group on Nutrition in Lysosomal Storage Diseases. Nutrition in adult patients with selected lysosomal storage diseases. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 733–744. [Google Scholar] [CrossRef]
- Luca, A.C.; David, S.G.; David, A.G.; Țarcă, V.; Pădureț, I.-A.; Mîndru, D.E.; Roșu, S.T.; Roșu, E.V.; Adumitrăchioaiei, H.; Bernic, J.; et al. Atherosclerosis from Newborn to Adult—Epidemiology, Pathological Aspects, and Risk Factors. Life 2023, 13, 2056. [Google Scholar] [CrossRef]
- Abumansour, I.S.; Yuskiv, N.; Paschke, E.; Stockler-Ipsiroglu, S. Morquio-B disease: Clinical and genetic characteristics of a distinct GLB1-related dysostosis multiplex. JIMD Rep. 2019, 51, 30–44. [Google Scholar] [CrossRef]
- Lee, J.S.; Choi, J.M.; Lee, M.; Kim, S.Y.; Lee, S.; Lim, B.C.; Cheon, J.E.; Kim, I.O.; Kim, K.J.; Choi, M.; et al. Diagnostic challenge for the rare lysosomal storage disease: Late infantile GM1 gangliosidosis. Brain Dev. 2018, 40, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.C.; Lozneanu, L.; Miron, I.C.; Trandafir, L.M.; Cojocaru, E.; Pădureţ, I.A.; Mihăilă, D.; Leon-Constantin, M.M.; Chiriac, Ş.; Iordache, A.C.; et al. Endocardial fibroelastosis and dilated cardiomyopathy—The past and future of the interface between histology and genetics. Romanian J. Morphol. Embryol. 2021, 61, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, M.P.; Iacobellis, F.; Acampora, E.; Caiazza, M.; Rubino, M.; Monda, E.; Magaldi, M.R.; Tarallo, A.; Sasso, M.; De Pasquale, V.; et al. Aortopathies in mouse models of Pompe, Fabry and Mucopolysaccharidosis IIIB lysosomal storage diseases. PLoS ONE 2020, 15, e0233050. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Belanger, E.C.; Veinot, J.P. Lysosomal storage disorders affecting the heart: A review. Cardiovasc. Pathol. 2019, 39, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Duceac, L.D.; Marcu, C.; Ichim, D.L.; Ciomaga, I.M.; Tarca, E.; Iordache, A.C.; Ciuhodaru, M.I.; Florescu, L.; Tutunaru, D.; Luca, A.C.; et al. Antibiotic Molecules Involved in Increasing Microbial Resistance. Rev. Chim. 2019, 70, 2622–2626. [Google Scholar] [CrossRef]
- Sinigerska, I.; Chandler, D.; Vaghjiani, V.; Hassanova, I.; Gooding, R.; Morrone, A.; Kremensky, I.; Kalaydjieva, L. Founder mutation causing infantile GM1-gangliosidosis in the Gypsy population. Mol. Genet. Metab. 2006, 88, 93–95. [Google Scholar] [CrossRef]
- Santos, R.; Amaral, O. Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy. Int. J. Mol. Sci. 2019, 20, 5897. [Google Scholar] [CrossRef]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Hum. Gene Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef]
- Gupta, M.; Pandey, H.; Sivakumar, S. Intracellular delivery of beta-galactosidase enzyme using arginase-responsive dextran sulfate/Poly-l-arginine capsule for lysosomal storage disorder. ACS Omega 2017, 2, 9002–9012. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.M.; Gross, A.L.; Martin, D.R.; Byrne, M.E. Polyethylene glycol-b-poly (lactic acid) polymersomes as vehicles for enzyme replacement therapy. Nanomedicine 2017, 12, 2591–2606. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Tang, Y.; Gandotra, N.; Enns, G.M.; Cowan, T.M.; Zhao, H.; Scharfe, C. Ethnic variability in newborn metabolic screening markers associated with false-positive outcomes. J. Inherit. Metab. Dis. 2020, 43, 934–943. [Google Scholar] [CrossRef]
- Țarcă, V.; Țarcă, E.; Luca, F.A. The Impact of the Main Negative Socio-Economic Factors on Female Fertility. Healthcare 2022, 10, 734. [Google Scholar] [CrossRef]
- Fritz, C.D.; Khan, J.; Kontoyiannis, P.D.; Cao, E.M.; Lawrence, A.; Love, L.D. Analysis of a Community Health Screening Program and the Factors Affecting Access to Care. Cureus 2023, 15, e41907. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gangliosidosis Type I | Morquio Type B (MBD) | Mucopolysaccharidosis (Except MBD) | Glycoproteinosis | |||
|---|---|---|---|---|---|---|
| Age of Onset | Infantile | Juvenile | 10–30 Years | 3–5 Years | Infantile/Juvenile | Infantile/ Juvenile/Adult |
| Facial features | Coarse | Mildly coarse | Dysmorphic or normal | - | Coarse | Coarse |
| Ocular | CRS | CC | CC | CC | CC; Glaucoma; Retinal dystrophy | CRS/CC |
| Skin | Mongolian spots | - | - | - | - | Angiokeratoma; Telangiectasia |
| Neurologic | EDD; Hypotonia Deafness; Blindness; Exaggerated startle response | Motor and speech impairment | Dystonia; Extrapiramidal signs | Normal | Hearing loss | Hearing loss; Ataxia; Seizures; Spasticity |
| Respiratory | Restrictive lung disease | - | - | Restrictive lung disease | Upper airway obstruction; Restrictive lung disease | - |
| Cardiovascular | HCM/DCM | HCM/DCM | HCM/DCM | Valvular defects | Valvular defects | Cardiomyopathies; Valvular anomalies |
| Digestive | Hepatosplenomegaly | Hepatosplenomegaly | - | Liver dysfunction | Hepatosplenomegaly; Diarrhea; Swallowing difficulties | Hepatosplenomegaly |
| Hematologic | Vacuolated lymphocytes | Vacuolated lymphocytes | Vacuolated lymphocytes | - | Anemia, thrombocytopenia, and coagulopathy in some diseases | Vacuolated lymphocytes; Immunodeficiency |
| Motor | + | + | + | - | - | + |
| Skeletal | Scoliosis; Dysostosis multiplex | Variable | Short stature | Dysostosis multiplex; Genu valgus; Short stature | Joint stiffness; Short stature | Dysostosis multiplex; Short stature |
| Renal | - | - | - | - | UTI and renal dysfunction possible | Renal failure |
| Cognitive | Severe impairment | Progressive decline | Intellectual disability | Normal intellect | Different stages of Intellectual disability | Progressive decline; Autism spectrum disorder |
| Non-genetic diagnostic tests | Urinary oligosaccharides; Elevated proinflammatory citokines; Enzyme activity | Urinary oligosaccharides; Elevated proinflammatory citokines; Enzyme activity | Urinary oligosaccharides; Elevated pro inflammatory citokines; Enzyme activity | Urinary excretion of KS by LC-MS; Elevated proinflammatory citokines; Enzyme activity | Urinary excretion of glycosaminoglycans by LC-MS; Enzyme activity | Urinary sialic acid-rich oligosaccharides; Enzyme activity |
| Genetic tests | GLB1 | GLB1 | GLB1 | GLB1 | Monogenic disorders | Monogenic disorders |
| Treatment options | Supportive and symptomatic; PC are under investigations | Supportive and symptomatic; PC are under investigations | Supportive and symptomatic; PC are under investigations | Supportive and symptomatic; PC are under investigations | ERT does not improve CNS symptoms; HSC transplant | Supportive and symptomatic; ERT available for alfa-Mannosidosis |
| Dietary strategies | Parenteral feeding/Enteral feeding using G-tubes | Ketogenic diet + Miglustat | Ketogenic diet + Miglustat | Antioxidants; Vitamin B6; Limited intake of sugar and milk | Oral zinc therapy in α-mannosidosis | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mîndru, D.E.; Țarcă, E.; Braha, E.E.; Curpăn, A.-Ș.; Roșu, S.T.; Anton-Păduraru, D.-T.; Adumitrăchioaiei, H.; Bernic, V.; Pădureț, I.-A.; Luca, A.C. Congenital Heart Malformations Masked by Infantile Gangliosidosis—Case Report and Growing Evidence for Metabolic Disease-Associated Aortopathies. Diagnostics 2024, 14, 491. https://doi.org/10.3390/diagnostics14050491
Mîndru DE, Țarcă E, Braha EE, Curpăn A-Ș, Roșu ST, Anton-Păduraru D-T, Adumitrăchioaiei H, Bernic V, Pădureț I-A, Luca AC. Congenital Heart Malformations Masked by Infantile Gangliosidosis—Case Report and Growing Evidence for Metabolic Disease-Associated Aortopathies. Diagnostics. 2024; 14(5):491. https://doi.org/10.3390/diagnostics14050491
Chicago/Turabian StyleMîndru, Dana Elena, Elena Țarcă, Elena Emanuela Braha, Alexandrina-Ștefania Curpăn, Solange Tamara Roșu, Dana-Teodora Anton-Păduraru, Heidrun Adumitrăchioaiei, Valentin Bernic, Ioana-Alexandra Pădureț, and Alina Costina Luca. 2024. "Congenital Heart Malformations Masked by Infantile Gangliosidosis—Case Report and Growing Evidence for Metabolic Disease-Associated Aortopathies" Diagnostics 14, no. 5: 491. https://doi.org/10.3390/diagnostics14050491
APA StyleMîndru, D. E., Țarcă, E., Braha, E. E., Curpăn, A.-Ș., Roșu, S. T., Anton-Păduraru, D.-T., Adumitrăchioaiei, H., Bernic, V., Pădureț, I.-A., & Luca, A. C. (2024). Congenital Heart Malformations Masked by Infantile Gangliosidosis—Case Report and Growing Evidence for Metabolic Disease-Associated Aortopathies. Diagnostics, 14(5), 491. https://doi.org/10.3390/diagnostics14050491