
Exome Sequencing Reveals Biallelic Mutations in MBTPS1 Gene in a Girl with a Very Rare Skeletal Dysplasia

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Extraction and Sequencing
2.2. Bioinformatics Pipeline, Variant Detection and Interpretation
3. Results
3.1. Case Report
3.2. Bioinformatic Analysis Revealed Pathogenic Compound Heterozygous Mutation in MBTPS1 Gene
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kondo, Y.; Fu, J.; Wang, H.; Hoover, C.; McDaniel, J.M.; Steet, R.; Patra, D.; Song, J.; Pollard, L.; Cathey, S.; et al. Site-1 protease deficiency causes human skeletal dysplasia due to defective inter-organelle protein trafficking. Clin. Investig. 2018, 3, e121596. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Elbracht, M.; Opladen, T.; Eggermann, T. Patient with an autosomal-recessive MBTPS1-linked phenotype and clinical features of Silver-Russell syndrome. Am. J. Med. Genet. A 2020, 182, 2727–2730. [Google Scholar] [CrossRef]
- Wang, X.; Sato, R.; Brown, M.S.; Hua, X.; Goldstein, J.L. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 1994, 77, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Marschner, K.; Kollmann, K.; Schweizer, M.; Braulke, T.; Pohl, S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science 2011, 333, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Steet, R.A.; Hullin, R.; Kudo, M.; Martinelli, M.; Bosshard, N.U.; Schaffner, T.; Kornfeld, S.; Steinmann, B. A splicing mutation in the ?/? GlcNAc-1-phosphotransferase gene results in an adult onset form of mucolipidosis III associated with sensory neuropathy and cardiomyopathy. Am. J. Med. Genet. Part A 2005, 132A, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Goldstein, J.L.; Hammer, R.E.; Moon, Y.-A.; Brown, M.S.; Horton, J.D. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc. Natl. Acad. Sci. USA 2001, 98, 13607–13612. [Google Scholar] [CrossRef] [PubMed]
- Schlombs, K.; Wagner, T.; Scheel, J. Site-1 protease is required for cartilage development in zebrafish. Proc. Natl. Acad. Sci. USA 2003, 100, 14024–14029. [Google Scholar] [CrossRef]
- Achilleos, A.; Huffman, N.T.; Marcinkiewicyz, E.; Seidah, N.G.; Chen, Q.; Dallas, S.L.; Trainor, P.A.; Gorski, J.P. MBTPS1/SKI-1/S1P proprotein convertase is required for ECM signaling and axial elongation during somitogenesis and vertebral development. Hum. Mol. Genet. 2015, 24, 2884–2898. [Google Scholar] [CrossRef]
- Patra, D.; Xing, X.; Davies, S.; Bryan, J.; Franz, C.; Hunziker, E.B.; Sandell, L.J. Site-1 protease is essential for endochondral bone formation in mice. J. Cell Biol. 2007, 179, 687–700. [Google Scholar] [CrossRef]
- Plaiasu, V.; Nanu, M.; Matei, D. Rare Disease Day-at a glance. Maedica 2010, 5, 65–66. [Google Scholar]
- Yuan, Y.; Zhou, Q.; Wang, C.; Zhou, W.; Gu, W.; Zheng, B. Clinical and molecular characterization of a patient with MBTPS1 related spondyloepiphyseal dysplasia: Evidence of pathogenicity for a synonymous variant. Front. Pediatr. 2022, 10, 1056141. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, M.; Aldossari, A.; Khan, I.; Alotaibi, L. Identification of a New Variant of the MBTPS1 Gene of the Kondo-Fu Type of Spondyloepiphyseal Dysplasia (SEDKF) in a Saudi Patient. Case Rep. Pediatr. 2022, 2022, 5498109. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.R.; Speck-Martins, C.E.; Brum, J.M.; Ferreira, C.R.; Sobreira, N.L.M. Spondyloepimetaphyseal dysplasia with elevated plasma lysosomal enzymes caused by homozygous variant in MBTPS1. Am. J. Med. Genet. A 2020, 182, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics-FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 July 2023).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Hu, J.; Ng, P.C. SIFT Indel: Predictions for the functional effects of amino acid insertions/deletions in proteins. PLoS ONE 2013, 8, e77940. [Google Scholar] [CrossRef]
- Schweitzer, G.G.; Gan, C.; Bucelli, R.C.; Wegner, D.; Schmidt, R.E.; Shinawi, M.; Finck, B.N.; Brookheart, R.T. A mutation in Site-1 Protease is associated with a complex phenotype that includes episodic hyperCKemia and focal myoedema. Mol. Genet. Genomic. Med. 2019, 7, e00733. [Google Scholar] [CrossRef]
- Chen, F.; Ni, C.; Wang, X.; Cheng, R.; Pan, C.; Wang, Y.; Liang, J.; Zhang, J.; Cheng, J.; Chin, Y.E.; et al. S1P defects cause a new entity of cataract, alopecia, oral mucosal disorder, and psoriasis-like syndrome. EMBO Mol. Med. 2022, 14, e14904. [Google Scholar] [CrossRef]
- Chen, C.; Wu, J.; Liu, Y. Case Report: Recombinant human growth hormone therapy in a patient with spondyloepiphyseal dysplasia, Kondo-Fu type. Front. Pediatr. 2023, 11, 1068718. [Google Scholar] [CrossRef]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Yuji Kondo et al., 2018 [1] | Schweitzer et al., 2019 [20] | Meyer et al., 2020 [2] | Carvalho et al., 2020 [13] | Chen et al., 2022 (Patient 1) [21] | Chen et al., 2022 (Patient 2) [21] | Alotaibi et al., 2022 [12] | Yuan et al., 2022 [11] | Chen et al., 2023 [22] | Our Case | YES % | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PMID: 30046013 | PMID: 31070020 | PMID: 32857899 | PMID: 32420688 | PMID: 35362222 | PMID: 35362222 | PMID: 36330313 | PMID: 36714646 | PMID: 36816387 | NA | ||
| Sex | F | F | M | F | M | F | F | M | M | F | 60 (F) |
| Age (years) | 8 | 24 | 7 | 5 | 14 | 5 | 10 | 12 | 6 | 4 | |
| Small for gestational age | Yes | No | Yes | Yes | NA | NA | Yes | No | No | Yes | 50 |
| Short stature | Yes | No | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | 80 |
| Low weight | Yes | No | Yes | Yes | NA | NA | Yes | Yes | Yes | Yes | 70 |
| Spondyloepiphyseal dysplasia on X-ray | Yes | NA | No | Yes | Yes | No | Yes | Yes | Yes | Yes | 70 |
| Prominent forehead | Yes | No | No | Yes | No | No | Yes | No | Yes | Yes | 50 |
| Micrognathia | No | No | No | Yes | No | No | Yes | Yes | Yes | Yes | 50 |
| Large and/or dysplastic ears | Yes | No | Yes | Yes | No | No | Yes | Yes | Yes | Yes | 70 |
| Bilateral cataracts, early onset | Yes (2 yo) | No | Yes (adolescence) | Yes (10 mo) | Yes (?) | Yes (?) | No | Yes (2 yo) | Yes (2 yo) | Yes (2 yo) | 80 |
| Pectus carinatum | Yes | No | No | Yes | NA | NA | Yes (mild) | Yes | No | Yes | 50 |
| Pectus excavatum | No | No | No | No | NA | NA | No | No | Yes | No | 10 |
| Inguinal hernias | Yes | No | Yes | No | No | No | No | Yes | No | Yes | 40 |
| Low bone mineral density | No | No | No | Yes (radiography) | NA | NA | Yes | Yes (radiography) | Yes (radiography) | Not assessed | 40 |
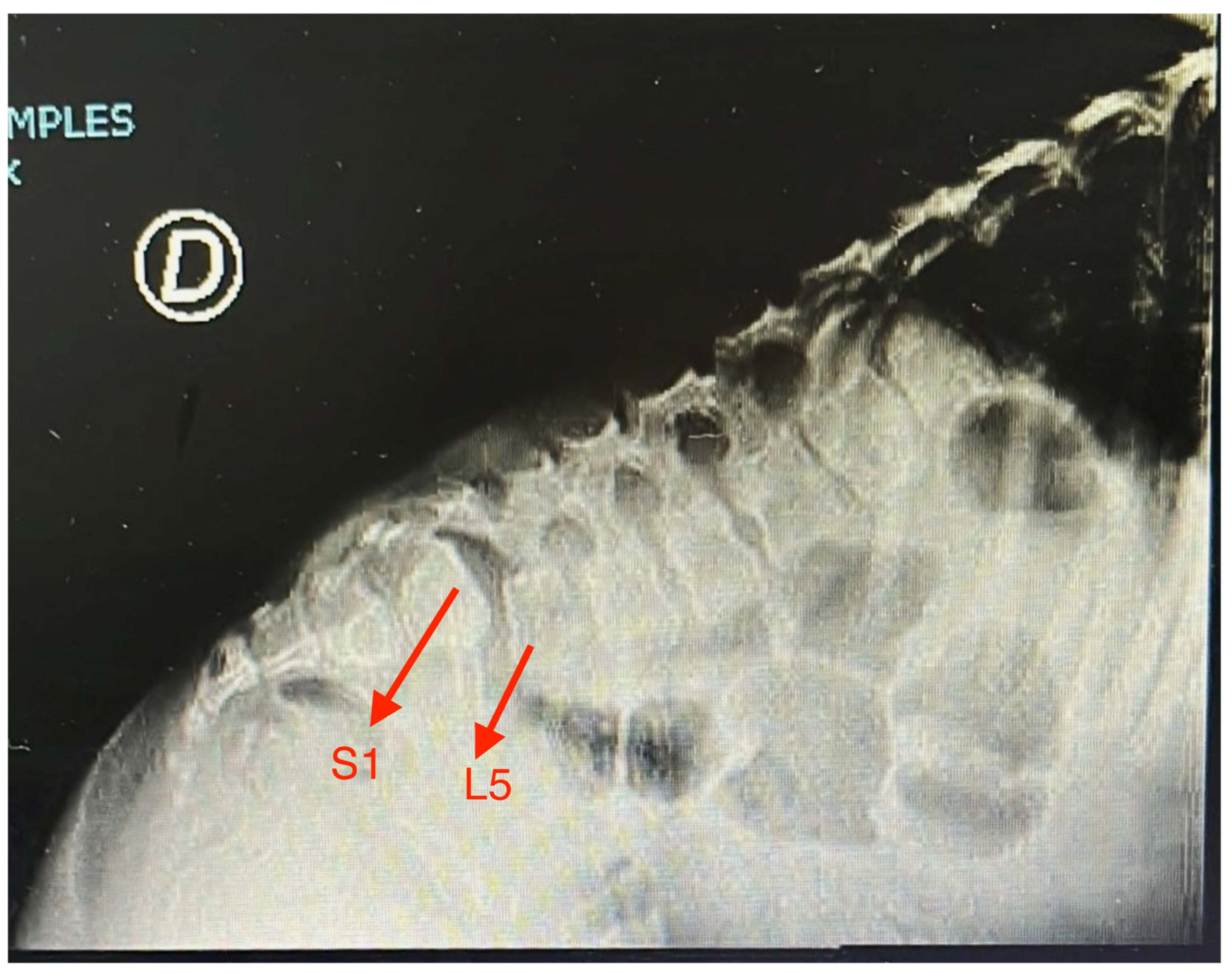
| Anterolisthesis of L5 on S1 | Yes | No | No | No | No | No | No | No | No | Yes | 20 |
| Kypho-scoliosis | Yes | No | No | Yes | NA | NA | Yes | Yes (mild) | Yes | No | 50 |
| Chronic back pain | Yes | No | No | No | No | No | No | No | No | Yes | 20 |
| Bilateral shortening of femoral necks | Yes | No | No | Yes | NA | NA | No | No | No | Yes | 30 |
| Irregular and dysplastic appearance of femoral epiphyses | Yes | No | No | Yes | NA | NA | Yes | Yes | No | Yes | 50 |
| Irregular and dysplastic appearance of proximal tibial epiphyses | Yes | No | No | Yes | NA | NA | No | No | No | No | 20 |
| Gracile fibulae | Yes | No | No | No | NA | NA | Yes | No | No | No | 20 |
| Valgus bowing of tibiae | Yes | No | No | No | NA | NA | Yes | No | No | No | 20 |
| Defective endochondral ossification | Yes | No | No | No | NA | NA | No | No | No | No | 10 |
| Delayed ossification of epiphyses | No | No | No | No | NA | NA | No | No | No | No | 0 |
| Delayed ossification of carpal bones | No | No | No | No | NA | NA | No | No | Yes | Not assessed | 10 |
| Brachydactyly | No | No | No | No | NA | NA | Yes | No | No | Yes | 20 |
| Shortening of tubular bones | No | No | No | Yes | NA | NA | Yes | No | No | Yes | 30 |
| Delayed gross motor milestones | Yes | No | No | Yes | No | Yes | Yes | No | No | No | 40 |
| Elevated lysosomal enzymes | Yes | No | Yes | Yes | No | No | Yes | Not assessed | Not assessed | Not assessed | 40 |
| HyperCKemia | No | Yes | No | No | No | No | No | No | No | No | 10 |
| Focal myoedema | No | Yes | No | No | No | No | No | No | No | No | 10 |
| Cutaneous lesions | No | No | No | No | Yes (psoriasis, ichthyosis) | Yes (psoriasis, ichthyosis) | No | No | No | No | 20 |
| Mucosal lesions | No | No | No | No | Yes | No | No | No | No | No | 10 |
| Sparse hair | No | No | No | No | Yes | Yes | No | No | No | Yes | 30 |
| Consanguinity | No | Not assessed | Yes (distantly related) | Yes (second degree cousins) | No | No | Yes (cousins) | No | No | No | 30 |
| Associated genetic variant(s) (NM_003791.3) | c.285dupT, p.(Asp96X)/c.1094A-G, p.(Asp365Gly) | c.3007C>T, p.(Pro1003Ser) | c.1094A>G, p.(Asp365Gly) | c.2948G>A, p.(Trp983ter) | c.1064T>G, p.(Val355Gly)/c.3157T>C, p.(Ter1053Arg) | c.2072-2A>T, p.(Ter1053Cys) | c.2634C>A, p.(Ser878Arg) | c.2656C>T, p.(Q886 *)/c.774C>T, p.(A258=) | c.1589A>G, p.(Asp530Gly)/c.163T > C, p.(Glu55Lys) | c.2355delG, p.(Met785fs)/c.1094A>G, p.(Asp365Gly) | |
| Zigocity/inheritance | compound heterozygous | heterozygous, de novo | Homozygous | Homozygous | compound heterozygous | compound heterozygous | Homozygous | compound heterozygous | compound heterozygous | compound heterozygous | |
| Splicing, frameshift or stop codon | 2 | 0 | 0 | 2 | 1 | 2 | 0 | 2 | 0 | 1 | |
| Missense | 0 | 1 | 2 | 0 | 1 | 0 | 2 | 0 | 2 | 1 | |
| Functional studies on variants | Yes | Yes | No | No | Yes | Yes | No | Yes | No | No | |
| Initially Misdiagnosed as Silver–Russell syndrome | Yes | No | Yes | No | No | No | No | No | No | Yes | 30 |
| Final Diagnosis | SEDKF | complex phenotype | SEDKF | SEDKF | CAOP | CAOP | SEDKF | SEDKF | SEDKF | SEDKF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raggio, V.; Rodríguez, S.; Feder, S.; Gueçaimburú, R.; Spangenberg, L. Exome Sequencing Reveals Biallelic Mutations in MBTPS1 Gene in a Girl with a Very Rare Skeletal Dysplasia. Diagnostics 2024, 14, 313. https://doi.org/10.3390/diagnostics14030313
Raggio V, Rodríguez S, Feder S, Gueçaimburú R, Spangenberg L. Exome Sequencing Reveals Biallelic Mutations in MBTPS1 Gene in a Girl with a Very Rare Skeletal Dysplasia. Diagnostics. 2024; 14(3):313. https://doi.org/10.3390/diagnostics14030313
Chicago/Turabian StyleRaggio, Víctor, Soledad Rodríguez, Sandra Feder, Rosario Gueçaimburú, and Lucía Spangenberg. 2024. "Exome Sequencing Reveals Biallelic Mutations in MBTPS1 Gene in a Girl with a Very Rare Skeletal Dysplasia" Diagnostics 14, no. 3: 313. https://doi.org/10.3390/diagnostics14030313
APA StyleRaggio, V., Rodríguez, S., Feder, S., Gueçaimburú, R., & Spangenberg, L. (2024). Exome Sequencing Reveals Biallelic Mutations in MBTPS1 Gene in a Girl with a Very Rare Skeletal Dysplasia. Diagnostics, 14(3), 313. https://doi.org/10.3390/diagnostics14030313