
The Multisystem Impact of Long COVID: A Comprehensive Review

 ,
, 
Abstract
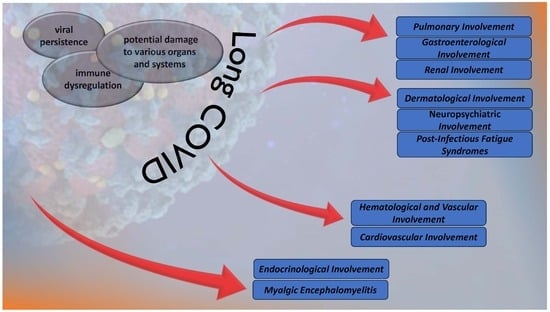
1. Introduction
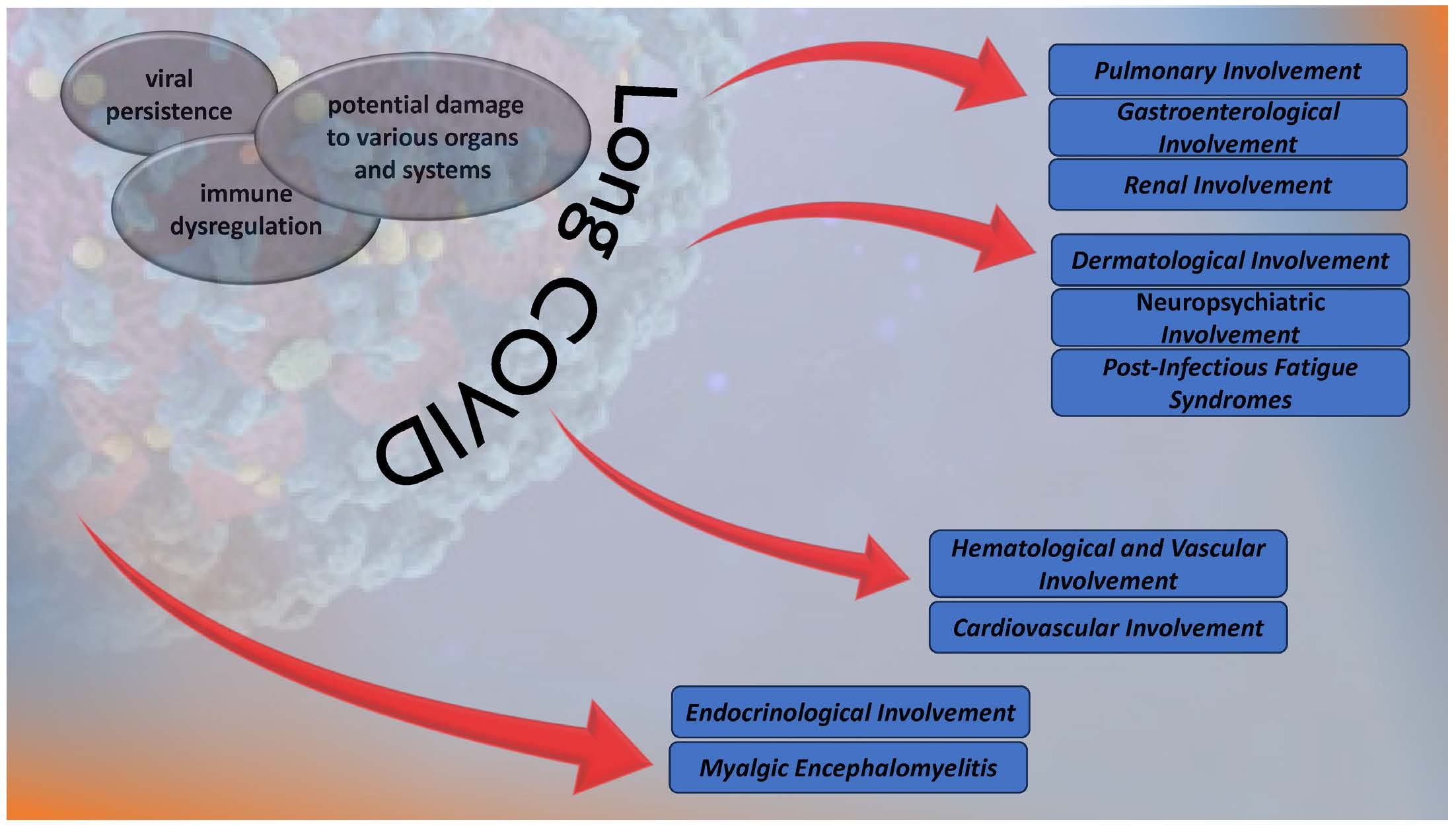
2. Materials and Methods
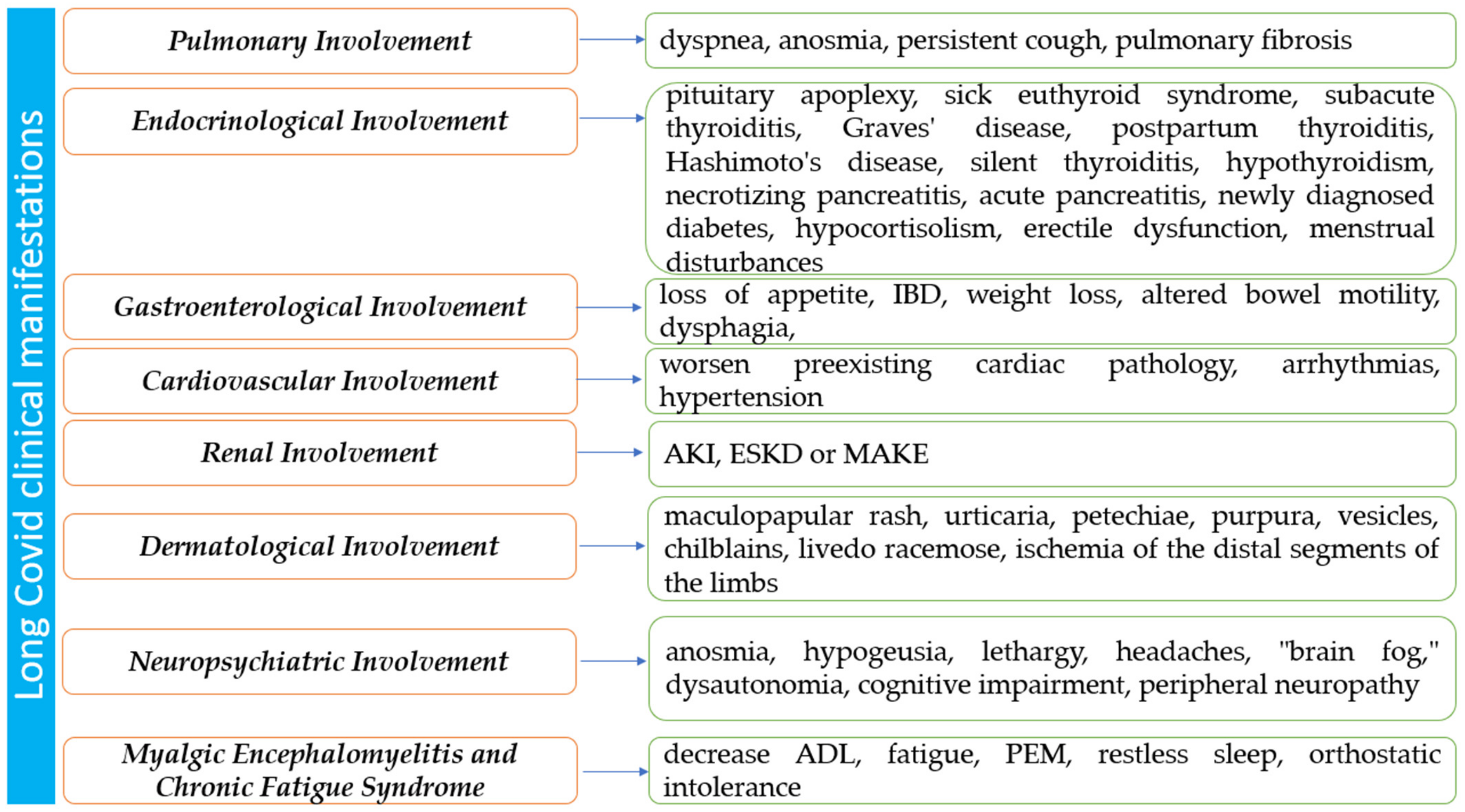
3. The Multisystem Impact of Long COVID
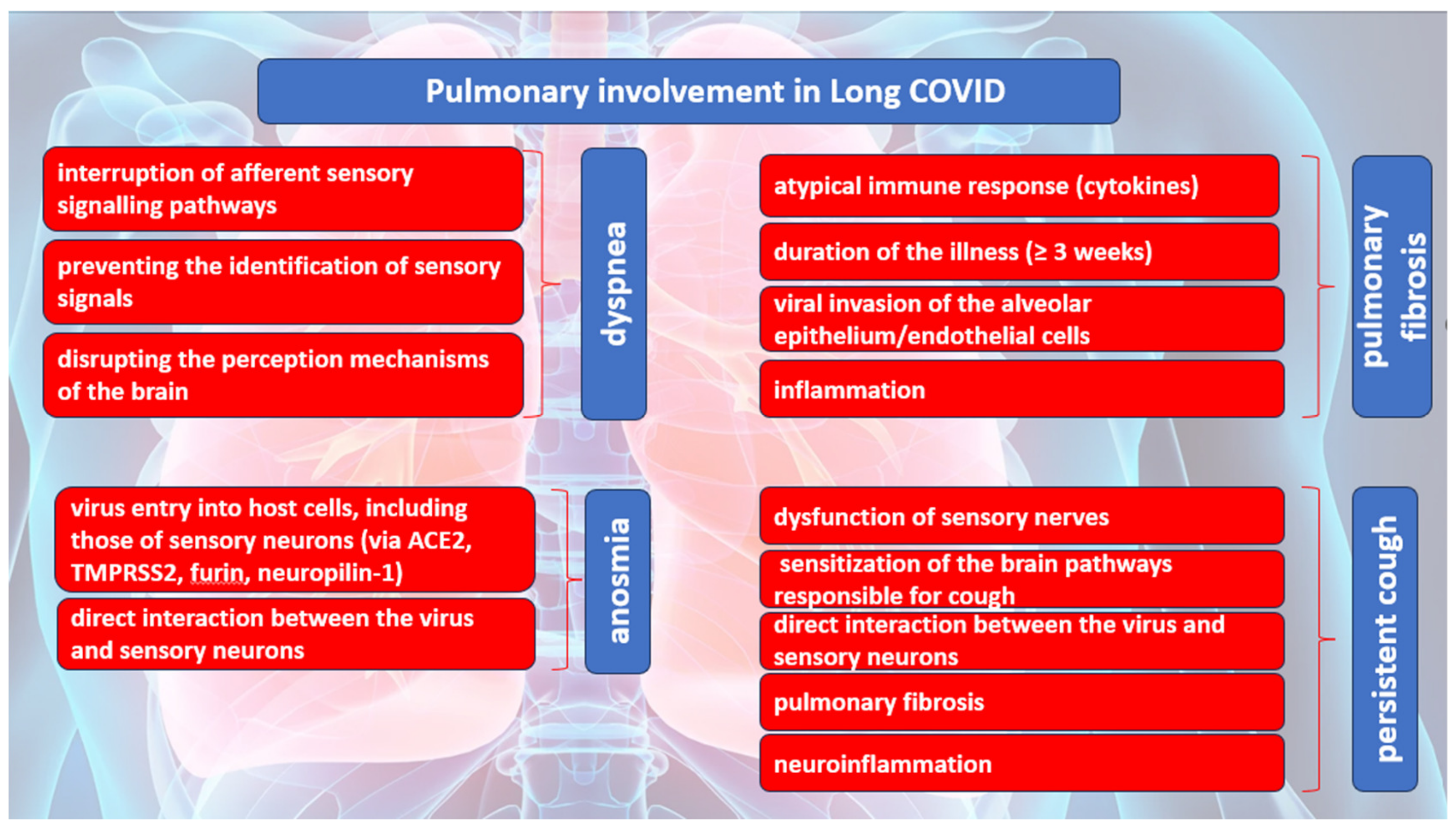
3.1. Pulmonary Involvement in Long COVID
3.2. Endocrinological Involvement in Long COVID
3.2.1. Pituitary Gland
3.2.2. Thyroid Gland
3.2.3. Pancreas
3.2.4. Adrenal Glands
3.2.5. Gonads
3.3. Hematological and Vascular Mechanism of Long COVID
3.3.1. Cell Count Alterations
3.3.2. SARS-CoV-2 Persistence in Extracellular Vesicles
3.3.3. Persistent Vascular Inflammatory State
3.3.4. Endothelial Damage and Dysfunction
3.3.5. Long-COVID-Related Hypercoagulability
3.3.6. Microangiopathy
3.4. Gastroenterological Involvement in Long COVID
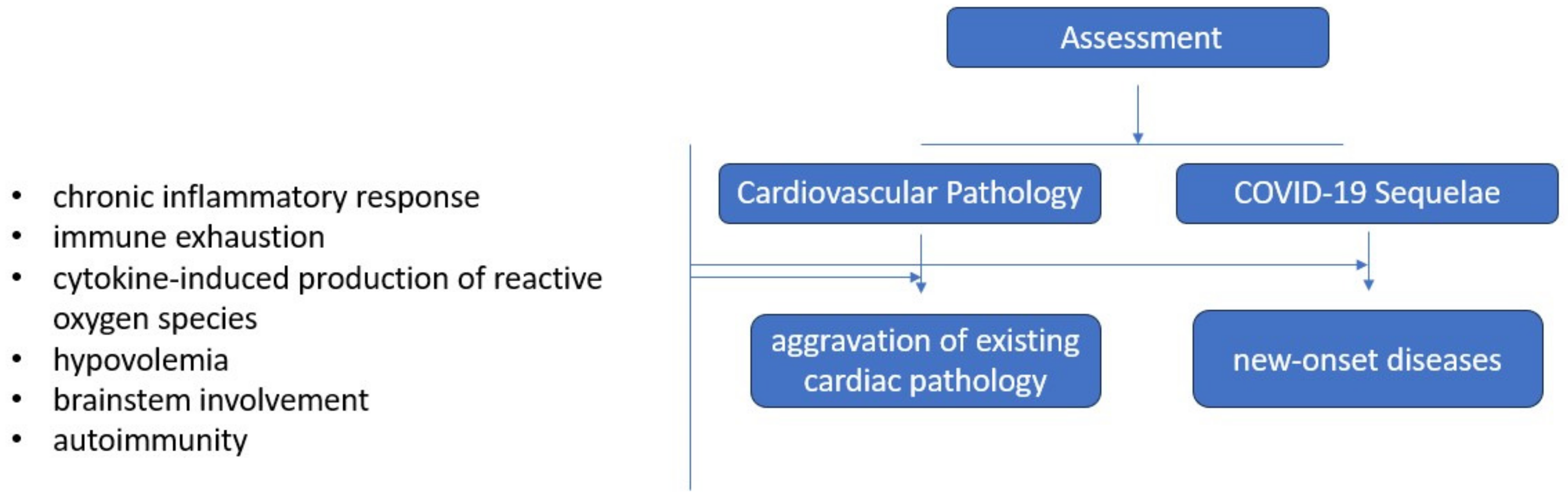
3.5. Cardiovascular Involvement in Long COVID
3.6. Renal Involvement in Long COVID
3.7. Dermatological Involvement in Long COVID
3.8. Neuropsychiatric Mechanism of Long COVID
3.8.1. SARS-CoV-2 Neurotropism
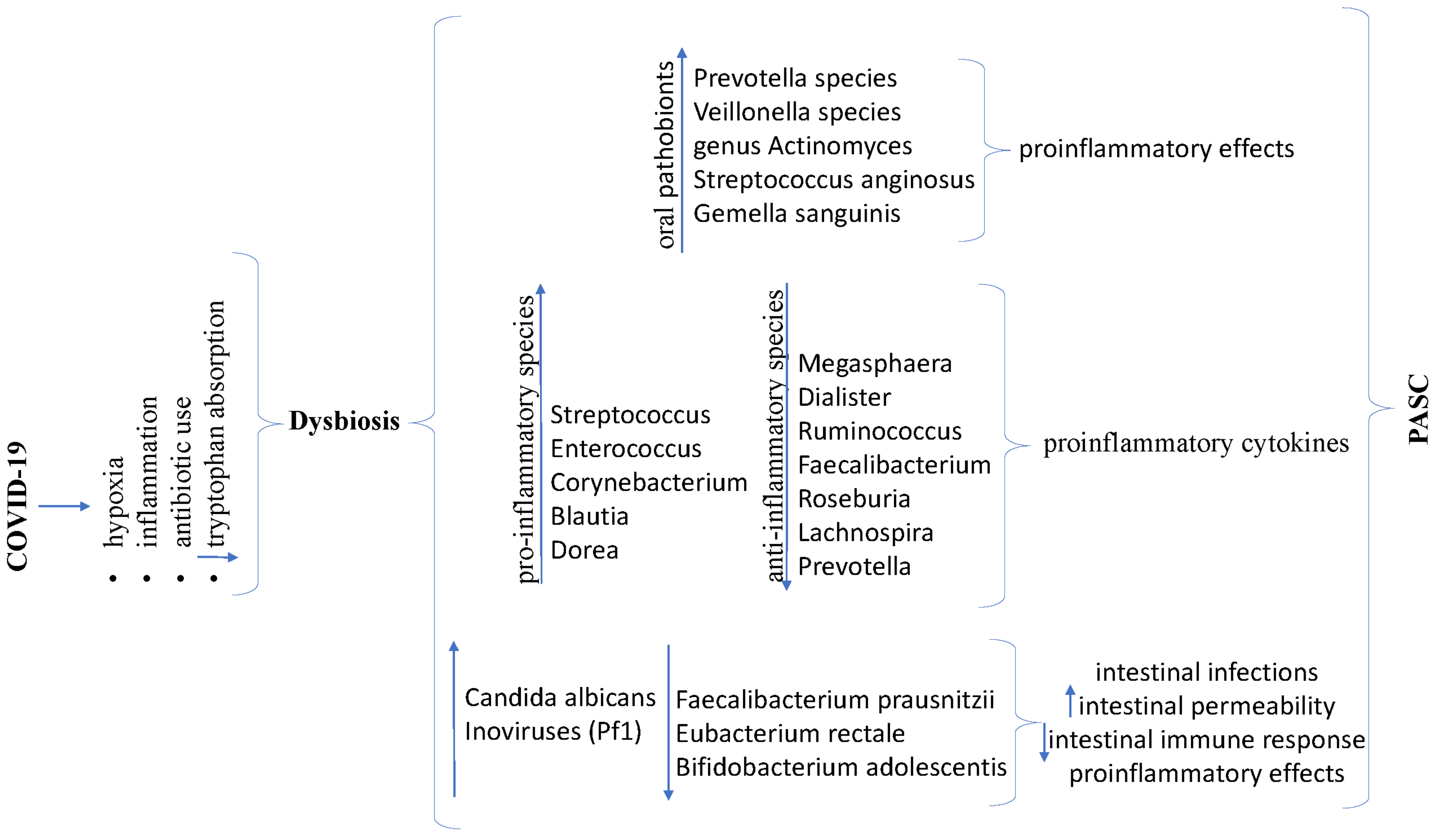
3.8.2. Brain–Gut Axis—Dysbiosis
3.8.3. Long COVID Neuroinflammation
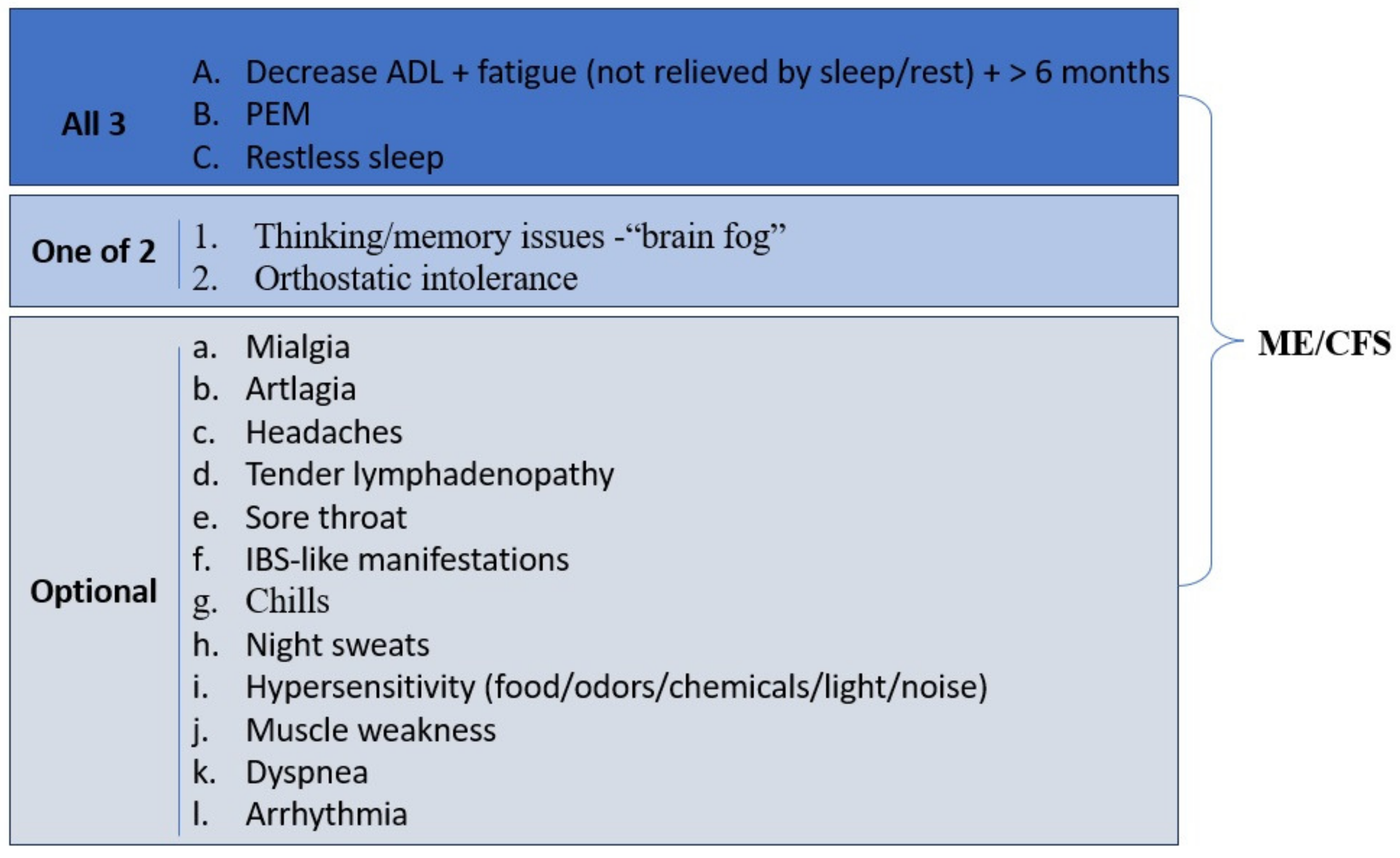
3.9. Myalgic Encephalomyelitis and Chronic Fatigue Syndrome in Long COVID-19
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soriano, J.B.; Murthy, S.; Marshall, J.C.; Relan, P.; Diaz, J.V. A clinical case definition of post-COVID-19 condition by a Delphi consensus. Lancet Infect. Dis. 2022, 22, e102–e107. [Google Scholar] [CrossRef] [PubMed]
- Sigfrid, L.; Drake, T.M.; Pauley, E.; Jesudason, E.C.; Olliaro, P.; Lim, W.S.; Gillesen, A.; Berry, C.; Lowe, D.J.; McPeake, J.; et al. Long COVID in Adults Discharged from UK Hospitals after COVID-19: A Prospective, Multicentre Cohort Study Using the ISARIC WHO Clinical Characterisation Protocol. Lancet Reg. Health-Eur. 2021, 8, 100186. [Google Scholar] [CrossRef] [PubMed]
- Desgranges, F.; Tadini, E.; Munting, A.; Regina, J.; Filippidis, P.; Viala, B.; Karachalias, E.; Suttels, V.; Haefliger, D.; Kampouri, E.; et al. Post-COVID-19 Syndrome in Outpatients: A Cohort Study. J. Gen. Intern. Med. 2022, 37, 1943–1952. [Google Scholar] [CrossRef]
- Subramanian, A.; Nirantharakumar, K.; Hughes, S.; Myles, P.; Williams, T.; Gokhale, K.M.; Taverner, T.; Chandan, J.S.; Brown, K.; Simms-Williams, N.; et al. Symptoms and Risk Factors for Long COVID in Non-Hospitalized Adults. Nat. Med. 2022, 28, 1706–1714. [Google Scholar] [CrossRef]
- Pelà, G.; Goldoni, M.; Solinas, E.; Cavalli, C.; Tagliaferri, S.; Ranzieri, S.; Frizzelli, A.; Marchi, L.; Mori, P.A.; Majori, M.; et al. Sex-Related Differences in Long-COVID-19 Syndrome. J. Women’s Health 2022, 31, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Gebhard, C.E.; Sütsch, C.; Bengs, S.; Todorov, A.; Deforth, M.; Buehler, K.P.; Meisel, A.; Schuepbach, R.A.; Zinkernagel, A.S.; Brugger, S.D.; et al. Understanding the Impact of Sociocultural Gender on Post-Acute Sequelae of COVID-19: A Bayesian Approach. MedRxiv 2021. [Google Scholar] [CrossRef]
- Tleyjeh, I.M.; Saddik, B.; Ramakrishnan, R.K.; AlSwaidan, N.; AlAnazi, A.; Alhazmi, D.; Aloufi, A.; AlSumait, F.; Berbari, E.F.; Halwani, R. Long Term Predictors of Breathlessness, Exercise Intolerance, Chronic Fatigue and Well-Being in Hospitalized Patients with COVID-19: A Cohort Study with 4 Months Median Follow-Up. J. Infect. Public Health 2022, 15, 21–28. [Google Scholar] [CrossRef]
- García-Abellán, J.; Padilla, S.; Fernández-González, M.; García, J.A.; Agulló, V.; Andreo, M.; Ruiz, S.; Galiana, A.; Gutiérrez, F.; Masiá, M. Antibody Response to SARS-CoV-2 Is Associated with Long-Term Clinical Outcome in Patients with COVID-19: A Longitudinal Study. J. Clin. Immunol. 2021, 41, 1490–1501. [Google Scholar] [CrossRef]
- Asadi-Pooya, A.A.; Akbari, A.; Emami, A.; Lotfi, M.; Rostamihosseinkhani, M.; Nemati, H.; Barzegar, Z.; Kabiri, M.; Zeraatpisheh, Z.; Farjoud-Kouhanjani, M.; et al. Risk Factors Associated with Long COVID Syndrome: A Retrospective Study. Iran. J. Med. Sci. 2021, 46, 428–436. [Google Scholar] [CrossRef]
- Munblit, D.; Bobkova, P.; Spiridonova, E.; Shikhaleva, A.; Gamirova, A.; Blyuss, O.; Nekliudov, N.; Bugaeva, P.; Andreeva, M.; DunnGalvin, A.; et al. Incidence and Risk Factors for Persistent Symptoms in Adults Previously Hospitalized for COVID-19. Clin. Exp. Allergy 2021, 51, 1107–1120. [Google Scholar] [CrossRef]
- Fernández-de-las-Peñas, C.; Martín-Guerrero, J.D.; Pellicer-Valero, Ó.J.; Navarro-Pardo, E.; Gómez-Mayordomo, V.; Cuadrado, M.L.; Arias-Navalón, J.A.; Cigarán-Méndez, M.; Hernández-Barrera, V.; Arendt-Nielsen, L. Female Sex Is a Risk Factor Associated with Long-Term Post-COVID Related-Symptoms but Not with COVID-19 Symptoms: The LONG-COVID-EXP-CM Multicenter Study. J. Clin. Med. 2022, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Tomasoni, D.; Falcinella, C.; Barbanotti, D.; Castoldi, R.; Mulè, G.; Augello, M.; Mondatore, D.; Allegrini, M.; Cona, A.; et al. Female Gender Is Associated with “Long COVID” Syndrome: A Prospective Cohort Study. Clin. Microbiol. Infect. 2021, 28, 611.e9–611.e16. [Google Scholar] [CrossRef] [PubMed]
- Chudzik, M.; Lewek, J.; Kapusta, J.; Banach, M.; Jankowski, P.; Bielecka-Dabrowa, A. Predictors of Long COVID in Patients without Comorbidities: Data from the Polish Long-COVID Cardiovascular (PoLoCOV-CVD) Study. J. Clin. Med. 2022, 11, 4980. [Google Scholar] [CrossRef]
- Chudzik, M.; Babicki, M.; Kapusta, J.; Kałuzińska-Kołat, Ż.; Kołat, D.; Jankowski, P.; Mastalerz-Migas, A. Long-COVID Clinical Features and Risk Factors: A Retrospective Analysis of Patients from the STOP-COVID Registry of the PoLoCOV Study. Viruses 2022, 14, 1755. [Google Scholar] [CrossRef] [PubMed]
- Notarte, K.I.; de Oliveira, M.H.S.; Peligro, P.J.; Velasco, J.V.; Macaranas, I.; Ver, A.T.; Pangilinan, F.C.; Pastrana, A.; Goldrich, N.; Kavteladze, D.; et al. Age, Sex and Previous Comorbidities as Risk Factors Not Associated with SARS-CoV-2 Infection for Long COVID-19: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 7314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-Converting Enzyme 2 (ACE2) as a SARS-CoV-2 Receptor: Molecular Mechanisms and Potential Therapeutic Target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.; Zuliani-Alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 Sensing by RIG-I and MDA5 Links Epithelial Infection to Macrophage Inflammation. EMBO J. 2021, 40, e107826. [Google Scholar] [CrossRef]
- Zhao, F.; Ma, Q.; Yue, Q.; Chen, H. SARS-CoV-2 Infection and Lung Regeneration. Clin. Microbiol. Rev. 2022, 35, e0018821. [Google Scholar] [CrossRef]
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. Expression of the SARS-CoV-2 ACE2 Receptor in the Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229. [Google Scholar] [CrossRef]
- Duclos, G.E.; Teixeira, V.H.; Autissier, P.; Gesthalter, Y.B.; Reinders-Luinge, M.A.; Terrano, R.; Dumas, Y.M.; Liu, G.; Mazzilli, S.A.; Brandsma, C.-A.; et al. Characterizing Smoking-Induced Transcriptional Heterogeneity in the Human Bronchial Epithelium at Single-Cell Resolution. Sci. Adv. 2019, 5, eaaw3413. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-Cell RNA-Seq Data Analysis on the Receptor ACE2 Expression Reveals the Potential Risk of Different Human Organs Vulnerable to 2019-NCoV Infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 Receptor ACE 2 and TMPRSS 2 Are Primarily Expressed in Bronchial Transient Secretory Cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Hentsch, L.; Cocetta, S.; Allali, G.; Santana, I.; Eason, R.; Adam, E.; Janssens, J.-P. Breathlessness and COVID-19: A Call for Research. Respiration 2021, 100, 1016–1026. [Google Scholar] [CrossRef]
- Coccia, C.B.I.; Palkowski, G.H.; Schweitzer, B.; Motsohi, T.; Ntusi, N. Dyspnoea: Pathophysiology and a Clinical Approach. S. Afr. Med. J. 2016, 106, 32. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, X.; Tong, Q.; Li, W.; Wang, B.; Sutter, K.; Trilling, M.; Lu, M.; Dittmer, U.; Yang, D. Overlapping and Discrete Aspects of the Pathology and Pathogenesis of the Emerging Human Pathogenic Coronaviruses SARS-CoV, MERS-CoV, and 2019-NCoV. J. Med. Virol. 2020, 92, 491–494. [Google Scholar] [CrossRef]
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.H.; Moss, M.; Downey, G.P. The Fibroproliferative Response in Acute Respiratory Distress Syndrome: Mechanisms and Clinical Significance. Eur. Respir. J. 2013, 43, 276–285. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 1031. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, H.; Zhou, Y.; Wu, X.; Zhao, Y.; Lu, Y.; Tan, W.; Yuan, M.; Ding, X.; Zou, J.; et al. Risk Factors Associated with Disease Severity and Length of Hospital Stay in COVID-19 Patients. J. Infect. 2020, 81, e95–e97. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.K.; Sharma, P.; Kumar, R. Post COVID 19 Pulmonary Fibrosis—Is It Reversible? Indian J. Tuberc. 2020, 68, 330–333. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary Fibrosis and COVID-19: The Potential Role for Antifibrotic Therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Valdes, A.M.; Freidin, M.B.; Sudre, C.H.; Nguyen, L.H.; Drew, D.A.; Ganesh, S.; Varsavsky, T.; Cardoso, M.J.; El-Sayed Moustafa, J.S.; et al. Real-Time Tracking of Self-Reported Symptoms to Predict Potential COVID-19. Nat. Med. 2020, 26, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, S.B.; Tian, L.; Moe, A.A.K.; Trewella, M.W.; Ritchie, M.E.; McGovern, A.E. Transcriptional Profiling of Individual Airway Projecting Vagal Sensory Neurons. Mol. Neurobiol. 2020, 57, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Randeva, H.; Chatha, K.; Hall, M.; Spandidos, D.; Karteris, E.; Kyrou, I. Neuropilin-1 as a New Potential SARS-CoV-2 Infection Mediator Implicated in the Neurologic Features and Central Nervous System Involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; den Berge, K.V.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-Neuronal Expression of SARS-CoV-2 Entry Genes in the Olfactory System Suggests Mechanisms Underlying COVID-19-Associated Anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef]
- Chen, M.; Shen, W.; Rowan, N.R.; Kulaga, H.; Hillel, A.; Ramanathan, M.; Lane, A.P. Elevated ACE-2 Expression in the Olfactory Neuroepithelium: Implications for Anosmia and Upper Respiratory SARS-CoV-2 Entry and Replication. Eur. Respir. J. 2020, 56, 2001948. [Google Scholar] [CrossRef]
- Shiers, S.; Ray, P.R.; Wangzhou, A.; Sankaranarayanan, I.; Tatsui, C.E.; Rhines, L.D.; Li, Y.; Uhelski, M.L.; Dougherty, P.M.; Price, T.J. ACE2 and SCARF Expression in Human Dorsal Root Ganglion Nociceptors: Implications for SARS-CoV-2 Virus Neurological Effects. Pain 2020, 161, 2494–2501. [Google Scholar] [CrossRef]
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 Protein of SARS-CoV-2 Crosses the Blood–Brain Barrier in Mice. Nat. Neurosci. 2020, 24, 368–378. [Google Scholar] [CrossRef]
- Ojha, V.; Mani, A.; Pandey, N.N.; Sharma, S.; Kumar, S. CT in Coronavirus Disease 2019 (COVID-19): A Systematic Review of Chest CT Findings in 4410 Adult Patients. Eur. Radiol. 2020, 30, 6129–6138. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Hilldrup, S.; Hope-Gill, B.D.; Eccles, R.; Harrison, N.K. Mechanical Induction of Cough in Idiopathic Pulmonary Fibrosis. Cough 2011, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.M.; Birring, S.S.; Gibson, P.G. Gabapentin for Refractory Chronic Cough: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2012, 380, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Vertigan, A.E.; Kapela, S.L.; Ryan, N.M.; Birring, S.S.; McElduff, P.; Gibson, P.G. Pregabalin and Speech Pathology Combination Therapy for Refractory Chronic Cough. Chest 2016, 149, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-Electron Microscopy Structures of the SARS-CoV Spike Glycoprotein Reveal a Prerequisite Conformational State for Receptor Binding. Cell Res. 2016, 27, 119–129. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Lazartigues, E.; Qadir, M.M.F.; Mauvais-Jarvis, F. Endocrine Significance of SARS-CoV-2’S Reliance on ACE2. Endocrinology 2020, 161, bqaa108. [Google Scholar] [CrossRef]
- Chan, J.L.; Gregory, K.D.; Smithson, S.S.; Naqvi, M.; Mamelak, A.N. Pituitary Apoplexy Associated with Acute COVID-19 Infection and Pregnancy. Pituitary 2020, 23, 716–720. [Google Scholar] [CrossRef]
- Ghosh, R.; Roy, D.; Roy, D.; Mandal, A.; Dutta, A.; Naga, D.; Benito-León, J. A Rare Case of SARS-CoV-2 Infection Associated with Pituitary Apoplexy without Comorbidities. J. Endocr. Soc. 2021, 5, bvaa203. [Google Scholar] [CrossRef] [PubMed]
- Bordes, S.J.; Phang-Lyn, S.; Najera, E.; Borghei-Razavi, H.; Adada, B. Pituitary Apoplexy Attributed to COVID-19 Infection in the Absence of an Underlying Macroadenoma or Other Identifiable Cause. Cureus 2021, 13, e13315. [Google Scholar] [CrossRef] [PubMed]
- Rotondi, M.; Coperchini, F.; Ricci, G.; Denegri, M.; Croce, L.; Ngnitejeu, S.T.; Villani, L.; Magri, F.; Latrofa, F.; Chiovato, L. Detection of SARS-CoV-2 Receptor ACE-2 MRNA in Thyroid Cells: A Clue for COVID-19-Related Subacute Thyroiditis. J. Endocrinol. Investig. 2020, 44, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Croce, L.; Gangemi, D.; Ancona, G.; Liboà, F.; Bendotti, G.; Minelli, L.; Chiovato, L. The Cytokine Storm and Thyroid Hormone Changes in COVID-19. J. Endocrinol. Investig. 2021, 44, 891–904. [Google Scholar] [CrossRef]
- Kumari, K.; Chainy, G.B.N.; Subudhi, U. Prospective Role of Thyroid Disorders in Monitoring COVID-19 Pandemic. Heliyon 2020, 6, e05712. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Chereji, A.-I.; Dodu, M.A.; Chereji, I.; Fitero, A.; Daina, C.M.; Daina, L.G.; Badau, D.; Neculoiu, D.C.; Domnariu, C. The Impact of Changes Regarding Working Circumstances during COVID-19 Pandemic upon Patients Evaluated for Thyroid Dysfunction. Int. J. Environ. Res. Public Health 2022, 19, 9856. [Google Scholar] [CrossRef]
- Fatourechi, V.; Aniszewski, J.P.; Fatourechi, G.Z.E.; Atkinson, E.J.; Jacobsen, S.J. Clinical Features and Outcome of Subacute Thyroiditis in an Incidence Cohort: Olmsted County, Minnesota, Study. J. Clin. Endocrinol. Metab. 2003, 88, 2100–2105. [Google Scholar] [CrossRef]
- Correia de Sá, T.; Soares, C.; Rocha, M. Acute Pancreatitis and COVID-19: A Literature Review. World J. Gastrointest. Surg. 2021, 13, 574–584. [Google Scholar] [CrossRef]
- Amidifar, S.; Elahi, R.; Siahmansouri, A.; Maleki, A.J.; Moradi, A. Endocrine and Metabolic Complications of COVID-19: Lessons Learned and Future Prospects. J. Mol. Endocrinol. 2022, 69, R125–R150. [Google Scholar] [CrossRef]
- Rubino, F.; Amiel, S.A.; Zimmet, P.; Alberti, G.; Bornstein, S.; Eckel, R.H.; Mingrone, G.; Boehm, B.; Cooper, M.E.; Chai, Z.; et al. New-Onset Diabetes in COVID-19. N. Engl. J. Med. 2020, 383, 789–790. [Google Scholar] [CrossRef]
- Landstra, C.P.; de Koning, E.J.P. COVID-19 and Diabetes: Understanding the Interrelationship and Risks for a Severe Course. Front. Endocrinol. 2021, 12, 649525. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, P.O. The Renin-Angiotensin System in the Endocrine Pancreas. JOP J. Pancreas 2001, 2, 26–32. [Google Scholar]
- Goossens, G.H. The Renin-Angiotensin System in the Pathophysiology of Type 2 Diabetes. Obes. Facts 2012, 5, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Karuparthi, P.R.; Habibi, J.; Wasekar, C.; Lastra, G.; Manrique, C.; Stas, S.; Sowers, J.R. Ultrastructural Islet Study of Early Fibrosis in the Ren2 Rat Model of Hypertension. Emerging Role of the Islet Pancreatic Pericyte-Stellate Cell. JOP J. Pancreas 2007, 8, 725–738. [Google Scholar]
- Wang, F.; Wang, H.; Fan, J.; Zhang, Y.; Wang, H.; Zhao, Q. Pancreatic Injury Patterns in Patients with COVID-19 Pneumonia. Gastroenterology 2020, 159, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Aloysius, M.M.; Thatti, A.; Gupta, A.; Sharma, N.; Bansal, P.; Goyal, H. COVID-19 Presenting as Acute Pancreatitis. Pancreatology 2020, 20, 1026–1027. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-Term Effects of COVID-19: A Systematic Review and Meta-Analysis. Medrxiv Prepr. Serv. Health Sci. 2021, 11, 16144. [Google Scholar] [CrossRef]
- Kamin, H.S.; Kertes, D.A. Cortisol and DHEA in Development and Psychopathology. Horm. Behav. 2017, 89, 69–85. [Google Scholar] [CrossRef]
- Wheatland, R. Molecular Mimicry of ACTH in SARS—Implications for Corticosteroid Treatment and Prophylaxis. Med. Hypotheses 2004, 63, 855–862. [Google Scholar] [CrossRef]
- Chu, K.Y.; Nackeeran, S.; Horodyski, L.; Masterson, T.A.; Ramasamy, R. COVID-19 Infection Is Associated with New Onset Erectile Dysfunction: Insights from a National Registry. Sex. Med. 2022, 10, 100478. [Google Scholar] [CrossRef]
- Barragan, M.; Guillén, J.J.; Martin-Palomino, N.; Rodriguez, A.; Vassena, R. Undetectable Viral RNA in Oocytes from SARS-CoV-2 Positive Women. Hum. Reprod. 2020, 36, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, G.; Hou, H.; Liao, Q.; Chen, J.; Bai, H.; Lee, S.; Wang, C.; Li, H.; Cheng, L.; et al. Analysis of Sex Hormones and Menstruation in COVID-19 Women of Child-Bearing Age. Reprod. BioMed. Online 2021, 42, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-Acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef] [PubMed]
- Report: What Does COVID-19 Recovery Actually Look Like? Patient Led Research. Available online: https://patientresearchcovid19.com/research/report-1/ (accessed on 20 November 2023).
- Erdinc, B.; Sahni, S.; Gotlieb, V. Hematological Manifestations and Complications of COVID-19. Adv. Clin. Exp. Med. 2021, 30, 101–107. [Google Scholar] [CrossRef]
- Sava, C.N.; Bodog, T.-M.; Niulas, L.R.; Iuhas, A.R.; Marinau, C.P.; Negrut, N.; Balmos, A.B.; Pasca, B.; Roman, N.A.; Delia Nistor-Cseppento, C. Biomarker Changes in Pediatric Patients with COVID-19: A Retrospective Study from a Single Center Database. Vivo 2022, 36, 2813–2822. [Google Scholar] [CrossRef] [PubMed]
- Harky, A.; Ala’Aldeen, A.; Butt, S.; Duric, B.; Roy, S.; Zeinah, M. COVID-19 and Multiorgan Response: The Long-Term Impact. Curr. Probl. Cardiol. 2023, 48, 101756. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets contribute to disease severity in COVID-19. J. Thromb. Haemost. 2021, 19, 3139–3153. [Google Scholar] [CrossRef] [PubMed]
- Targosz-Korecka, M.; Kubisiak, A.; Kloska, D.; Kopacz, A.; Grochot-Przeczek, A.; Szymonski, M. Endothelial Glycocalyx Shields the Interaction of SARS-CoV-2 Spike Protein with ACE2 Receptors. Sci. Rep. 2021, 11, 12157. [Google Scholar] [CrossRef]
- Prasad, M.; Leon, M.; Lerman, L.O.; Lerman, A. Viral Endothelial Dysfunction: A Unifying Mechanism for COVID-19. Mayo Clin. Proc. 2021, 96, 3099–3108. [Google Scholar] [CrossRef]
- Oikonomou, E.; Souvaliotis, N.; Lampsas, S.; Siasos, G.; Poulakou, G.; Theofilis, P.; Papaioannou, T.G.; Haidich, A.-B.; Tsaousi, G.; Ntousopoulos, V.; et al. Endothelial Dysfunction in Acute and Long Standing COVID−19: A Prospective Cohort Study. Vasc. Pharmacol. 2022, 144, 106975. [Google Scholar] [CrossRef]
- Chioh, F.W.; Fong, S.-W.; Young, B.E.; Wu, K.-X.; Siau, A.; Krishnan, S.; Chan, Y.-H.; Carissimo, G.; Teo, L.L.; Gao, F.; et al. Convalescent COVID-19 Patients Are Susceptible to Endothelial Dysfunction due to Persistent Immune Activation. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J.M.; et al. Persistent Endotheliopathy in the Pathogenesis of Long COVID Syndrome. J. Thromb. Haemost. 2021, 19, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Renteria, H.; Travieso, A.; Sagir, A.; Martínez-Gómez, E.; Carrascosa-Granada, A.; Toya, T.; Núñez-Gil, I.J.; Estrada, V.; Lerman, A.; Escaned, J. In-Vivo Evidence of Systemic Endothelial Vascular Dysfunction in COVID-19. Int. J. Cardiol. 2021, 345, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A.; Papatheou, D.; Melita, H. COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. J. Cardiovasc. Pharmacol. Ther. 2020, 26, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Poor, H.D. Pulmonary Thrombosis and Thromboembolism in COVID-19. Chest 2021, 160, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent Clotting Protein Pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) Is Accompanied by Increased Levels of Antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Kotze, M.J.; Oladejo, S.O.; Watson, L.R.; Rajaratnam, K.; Watson, B.W.; Kell, D.B. Prevalence of Symptoms, Comorbidities, Fibrin Amyloid Microclots and Platelet Pathology in Individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc. Diabetol. 2022, 21, 148. [Google Scholar] [CrossRef]
- Chen, W.; Pan, J.Y. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biol. Proced. Online 2021, 23, 4. [Google Scholar] [CrossRef]
- Guervilly, C.; Bonifay, A.; Burtey, S.; Sabatier, F.; Cauchois, R.; Abdili, E.; Arnaud, L.; Lano, G.; Pietri, L.; Robert, T.; et al. Dissemination of Extreme Levels of Extracellular Vesicles: Tissue Factor Activity in Patients with Severe COVID-19. Blood Adv. 2021, 5, 628–634. [Google Scholar] [CrossRef]
- Barberis, E.; Vanella, V.V.; Falasca, M.; Caneapero, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; De Giorgis, V.; Puricelli, C.; Vaschetto, R.; et al. Circulating Exosomes Are Strongly Involved in SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8, 29. [Google Scholar] [CrossRef]
- Eymieux, S.; Uzbekov, R.; Rouillé, Y.; Blanchard, E.; Hourioux, C.; Dubuisson, J.; Belouzard, S.; Roingeard, P. Secretory Vesicles Are the Principal Means of SARS-CoV-2 Egress. Cells 2021, 10, 2047. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.; Luque, N.; Blanco, I.; Sebastian, L.; Barberà, J.A.; Peinado, V.I.; Tura-Ceide, O. Pulmonary Endothelial Dysfunction and Thrombotic Complications in Patients with COVID-19. Am. J. Respir. Cell Mol. Biol. 2021, 64, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, C.; Jing, H.; Wu, X.; Novakovic, V.A.; Xie, R.; Shi, J. Long COVID: The Nature of Thrombotic Sequelae Determines the Necessity of Early Anticoagulation. Front. Cell. Infect. Microbiol. 2022, 12, 861703. [Google Scholar] [CrossRef]
- Sharma, P.; Behl, T.; Sharma, N.; Singh, S.; Grewal, A.S.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Bungau, S. COVID-19 and diabetes: Association intensify risk factors for morbidity and mortality. Biomed. Pharmacother. 2022, 151, 113089. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.T.; Uddin, M.S.; Hossain, M.F.; Abdulhakim, J.A.; Alam, M.A.; Ashraf, G.M.; Bungau, S.G.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Aleya, L. nCOVID-19 Pandemic: From Molecular Pathogenesis to Potential Investigational Therapeutics. Front. Cell Dev. Biol. 2020, 8, 616. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, G.; Ilie, M.; Bungau, S.; Stoian, A.M.P.; Bacalbasa, N.; Diaconu, C.C. Is There a Relationship between COVID-19 and Hyponatremia? Medicina 2021, 57, 55. [Google Scholar] [CrossRef] [PubMed]
- Tagde, P.; Tagde, S.; Tagde, P.; Bhattacharya, T.; Monzur, S.M.; Rahman, M.H.; Otrisal, P.; Behl, T.; ul Hassan, S.S.; Abdel-Daim, M.M.; et al. Nutraceuticals and Herbs in Reducing the Risk and Improving the Treatment of COVID-19 by Targeting SARS-CoV-2. Biomedicines 2021, 9, 1266. [Google Scholar] [CrossRef]
- Perumal, R.; Shunmugam, L.; Naidoo, K.; Wilkins, D.; Garzino-Demo, A.; Brechot, C.; Vahlne, A.; Nikolich, J. Biological Mechanisms Underpinning the Development of Long COVID. iScience 2023, 26, 106935. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Tokuyama, M.; Wei, G.; Huang, R.; Livanos, A.; Jha, D.; Levescot, A.; Irizar, H.; Kosoy, R.; Cording, S.; et al. Intestinal Inflammation Modulates the Expression of ACE2 and TMPRSS2 and Potentially Overlaps with the Pathogenesis of SARS-CoV-2–Related Disease. Gastroenterology 2021, 160, 287–301.e20. [Google Scholar] [CrossRef]
- Mehandru, S.; Merad, M. Pathological Sequelae of Long-Haul COVID. Nat. Immunol. 2022, 23, 194–202. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of Antibody Immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Tariq, R.; Jena, A.; Vesely, E.K.; Singh, S.; Khanna, S.; Sharma, V. Gastrointestinal Manifestations of Long COVID: A Systematic Review and Meta-Analysis. Ther. Adv. Gastroenterol. 2022, 15, 175628482211184. [Google Scholar] [CrossRef] [PubMed]
- Adame, M.J.; Nada, K.M.; Seashore, J. Late Sequelae of COVID-19 Infection in Patients without Comorbidities. Am. J. Respir. Crit. Care Med. 2021, 203, A3821. [Google Scholar]
- Ozyurek, B.A.; Ozdemirel, T.S.; Akkurt, E.S.; Yenibertiz, D.; Saymaz, Z.T.; Özden, S.B.; Eroğlu, Z. What Are the Factors That Affect Post COVID 1st Month’s Continuing Symptoms? Int. J. Clin. Pract. 2021, 75, e14778. [Google Scholar] [CrossRef]
- Faycal, A.; Ndoadoumgue, A.L.; Sellem, B.; Blanc, C.; Dudoit, Y.; Schneider, L.; Tubiana, R.; Valantin, M.-A.; Seang, S.; Palich, R.; et al. Prevalence and Factors Associated with Symptom Persistence: A Prospective Study of 429 Mild COVID-19 Outpatients. Infect. Dis. Now. 2022, 52, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fernández-de-las-Peñas, C.; Martín-Guerrero, J.; Navarro-Pardo, E.; Torres-Macho, J.; Canto-Diez, M.G.; Pellicer-Valero, O. Gastrointestinal Symptoms at the Acute COVID-19 Phase Are Risk Factors for Developing Gastrointestinal Post-COVID Symptoms: A Multicenter Study. Intern. Emerg. Med. 2021, 17, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Kozak, R.; Armstrong, S.M.; Salvant, E.; Ritzker, C.; Feld, J.; Biondi, M.J.; Tsui, H. Recognition of Long-COVID-19 Patients in a Canadian Tertiary Hospital Setting: A Retrospective Analysis of Their Clinical and Laboratory Characteristics. Pathogens 2021, 10, 1246. [Google Scholar] [CrossRef]
- Leth, S.; Gunst, J.D.; Mathiasen, V.D.; Hansen, K.S.; Søgaard, O.S.; Østergaard, L.; Jensen-Fangel, S.; Storgaard, M.; Agergaard, J. Persistent Symptoms in Hospitalized Patients Recovering from COVID-19 in Denmark. Open Forum Infect. Dis. 2021, 8, ofab042. [Google Scholar] [CrossRef]
- Lombardo, M.D.M.; Foppiani, A.; Peretti, G.M.; Mangiavini, L.; Battezzati, A.; Bertoli, S.; Martinelli Boneschi, F.; Zuccotti, G.V. Long-Term Coronavirus Disease 2019 Complications in Inpatients and Outpatients: A One-Year Follow-up Cohort Study. Open Forum Infect. Dis. 2021, 8, ofab384. [Google Scholar] [CrossRef]
- Shang, Y.F.; Liu, T.; Yu, J.N.; Xu, X.R.; Zahid, K.R.; Wei, Y.C.; Wang, X.H.; Zhou, F.L. Half-Year Follow-up of Patients Recovering from Severe COVID-19: Analysis of Symptoms and Their Risk Factors. J. Intern. Med. 2021, 290, 444–450. [Google Scholar] [CrossRef]
- Mouchati, C.; Durieux, J.C.; Zisis, S.N.; Labbato, D.; Rodgers, M.A.; Ailstock, K.; Reinert, B.L.; Funderburg, N.T.; McComsey, G.A. Increase in Gut Permeability and Oxidized Ldl Is Associated with Post-Acute Sequelae of SARS-CoV-2. Front. Immunol. 2023, 14, 1182544. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; van Schayck, J.P.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 Productively Infects Human Gut Enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Parker, J.; Smits, S.; Underwood, J.; Dolwani, S. Persistent viral shedding of SARS-CoV-2 in faeces—A rapid review. Color. Dis. 2020, 22, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.-H.; Ni, W.; Wu, Q.; Li, W.-J.; Li, G.-J.; Wang, W.-D.; Tong, J.-N.; Song, X.-F.; Wing-Kin Wong, G.; Xing, Q.-S. Prolonged Viral Shedding in Feces of Pediatric Patients with Coronavirus Disease 2019. J. Microbiol. Immunol. Infect. 2020, 53, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Amirian, E.S. Potential Fecal Transmission of SARS-CoV-2: Current Evidence and Implications for Public Health. Int. J. Infect. Dis. 2020, 95, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, L.; Deng, Q.; Zhang, G.; Wu, K.; Ni, L.; Yang, Y.; Liu, B.; Wang, W.; Wei, C.; et al. The Presence of SARS-CoV-2 RNA in the Feces of COVID-19 Patients. J. Med. Virol. 2020, 92, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Baluja, M.Q.; Graham, D.W.; Corbishley, A.; McDonald, J.E.; Malham, S.K.; Hillary, L.S.; Connor, T.R.; Gaze, W.H.; Moura, I.B.; et al. Shedding of SARS-CoV-2 in Feces and Urine and Its Potential Role in Person-To-Person Transmission and the Environment-Based Spread of COVID-19. Sci. Total Environ. 2020, 749, 141364. [Google Scholar] [CrossRef]
- Peng, L.; Liu, J.; Xu, W.; Luo, Q.; Chen, D.; Lei, Z.; Huang, Z.; Li, X.; Deng, K.; Lin, B.; et al. SARS-CoV-2 Can Be Detected in Urine, Blood, Anal Swabs, and Oropharyngeal Swabs Specimens. J. Med. Virol. 2020, 92, 1676–1680. [Google Scholar] [CrossRef]
- Tian, Y.; Rong, L.; Nian, W.; He, Y. Review Article: Gastrointestinal Features in COVID-19 and the Possibility of Faecal Transmission. Aliment. Pharmacol. Ther. 2020, 51, 843–851. [Google Scholar] [CrossRef]
- Khreefa, Z.; Barbier, M.T.; Koksal, A.R.; Love, G.; Del Valle, L. Pathogenesis and Mechanisms of SARS-CoV-2 Infection in the Intestine, Liver, and Pancreas. Cells 2023, 12, 262. [Google Scholar] [CrossRef]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological Dysfunction Persists for 8 Months Following Initial Mild-To-Moderate SARS-CoV-2 Infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, M.; Reistam, U.; Fedorowski, A.; Villacorta, H.; Horiuchi, Y.; Bax, J.; Pitt, B.; Matskeplishvili, S.; Lüscher, T.F.; Weichert, I.; et al. Post-COVID-19 Tachycardia Syndrome: A Distinct Phenotype of Post-Acute COVID-19 Syndrome. Am. J. Med. 2021, 134, 1451–1456. [Google Scholar] [CrossRef]
- Vernino, S.; Stiles, L.E. Autoimmunity in Postural Orthostatic Tachycardia Syndrome: Current Understanding. Auton. Neurosci. 2018, 215, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, G.; Ricci, F.; Recce, V.; Serio, A.; Iannetti, G.; Chahal, A.A.; Ståhlberg, M.; Khanji, M.Y.; Fedorowski, A.; Gallina, S. Post-Acute Sequelae of COVID-19 and Cardiovascular Autonomic Dysfunction: What Do We Know? J. Cardiovasc. Dev. Dis. 2021, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Taquet, M.; Dercon, Q.; Luciano, S.; Geddes, J.R.; Husain, M.; Harrison, P.J. Incidence, Co-Occurrence, and Evolution of Long-COVID Features: A 6-Month Retrospective Cohort Study of 273,618 Survivors of COVID-19. PLoS Med. 2021, 18, e1003773. [Google Scholar] [CrossRef] [PubMed]
- Schiffl, H.; Lang, S.M. Long-Term Interplay between COVID-19 and Chronic Kidney Disease. Int. Urol. Nephrol. 2023, 55, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Bowe, B.; Xie, Y.; Xu, E.; Al-Aly, Z. Kidney Outcomes in Long COVID. J. Am. Soc. Nephrol. 2021, 32, 2851–2862. [Google Scholar] [CrossRef]
- Chand, S.; Kapoor, S.; Naqvi, A.; Thakkar, J.; Fazzari, M.J.; Orsi, D.; Dieiev, V.; Lewandowski, D.C.; Dicpinigaitis, P.V. Long-Term Follow up of Renal and Other Acute Organ Failure in Survivors of Critical Illness due to COVID-19. J. Intensive Care Med. 2021, 37, 736–742. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A.B.; Nie, X.; et al. Renal Histopathological Analysis of 26 Postmortem Findings of Patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; et al. CD147-Spike Protein Is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Aleya, L.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci. Total. Environ. 2022, 808, 152072. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e907. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human Kidney Is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Sohier, P.; Benghanem, S.; L’Honneur, A.-S.; Rozenberg, F.; Dupin, N.; Garel, B. Digitate Papulosquamous Eruption Associated with Severe Acute Respiratory Syndrome Coronavirus 2 Infection. JAMA Dermatol. 2020, 156, 819. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, M.; Long, B. Dermatologic Manifestations and Complications of COVID-19. Am. J. Emerg. Med. 2020, 38, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Galván Casas, C.; Català, A.; Carretero Hernández, G.; Rodríguez-Jiménez, P.; Fernández Nieto, D.; Rodríguez-Villa Lario, A.; Navarro Fernández, I.; Ruiz-Villaverde, R.; Falkenhain, D.; Llamas Velasco, M.; et al. Classification of the Cutaneous Manifestations of COVID-19: A Rapid Prospective Nationwide Consensus Study in Spain with 375 Cases. Br. J. Dermatol. 2020, 183, 71–77. [Google Scholar] [CrossRef]
- Kaya, G.; Kaya, A.; Saurat, J.-H. Clinical and Histopathological Features and Potential Pathological Mechanisms of Skin Lesions in COVID-19: Review of the Literature. Dermatopathology 2020, 7, 3–16. [Google Scholar] [CrossRef]
- Suchonwanit, P.; Leerunyakul, K.; Kositkuljorn, C. Cutaneous Manifestations in COVID-19: Lessons Learned from Current Evidence. J. Am. Acad. Dermatol. 2020, 83, e57–e60. [Google Scholar] [CrossRef]
- Criado, P.R.; Abdalla, B.M.Z.; de Assis, I.C.; van Blarcum de Graaff Mello, C.; Caputo, G.C.; Vieira, I.C. Are the Cutaneous Manifestations during or due to SARS-CoV-2 Infection/COVID-19 Frequent or Not? Revision of Possible Pathophysiologic Mechanisms. Inflamm. Res. 2020, 69, 745–756. [Google Scholar] [CrossRef]
- Goren, A.; Vaño-Galván, S.; Wambier, C.G.; McCoy, J.; Gomez-Zubiaur, A.; Moreno-Arrones, O.M.; Shapiro, J.; Sinclair, R.D.; Gold, M.H.; Kovacevic, M.; et al. A Preliminary Observation: Male Pattern Hair Loss among Hospitalized COVID-19 Patients in Spain—A Potential Clue to the Role of Androgens in COVID-19 Severity. J. Cosmet. Dermatol. 2020, 19, 1545–1547. [Google Scholar] [CrossRef] [PubMed]
- Mahé, A.; Birckel, E.; Merklen, C.; Lefèbvre, P.; Hannedouche, C.; Jost, M.; Droy-Dupré, L. Histology of Skin Lesions Establishes That the Vesicular Rash Associated with COVID-19 Is Not “Varicella-Like”. J. Eur. Acad. Dermatol. Venereol. 2020, 34, E559–E561. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Leng, A.; Shah, M.; Ahmad, S.A.; Premraj, L.; Wildi, K.; Li Bassi, G.; Pardo, C.A.; Choi, A.; Cho, S.-M. Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics. Cells 2023, 12, 816. [Google Scholar] [CrossRef] [PubMed]
- Stefanou, M.-I.; Palaiodimou, L.; Bakola, E.; Smyrnis, N.; Papadopoulou, M.; Paraskevas, G.P.; Rizos, E.; Boutati, E.; Grigoriadis, N.; Krogias, C.; et al. Neurological Manifestations of Long-COVID Syndrome: A Narrative Review. Ther. Adv. Chronic Dis. 2022, 13, 20406223221076890. [Google Scholar] [CrossRef] [PubMed]
- Balcom, E.F.; Nath, A.; Power, C. Acute and Chronic Neurological Disorders in COVID-19: Potential Mechanisms of Disease. Brain 2021, 144, 3576–3588. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory Transmucosal SARS-CoV-2 Invasion as a Port of Central Nervous System Entry in Individuals with COVID-19. Nat. Neurosci. 2020, 24, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, Y.; Li, Y.; Wu, Q.; Wu, J.; Park, S.K.; Guo, C.; Lu, J. Meta-analysis of 16S rRNA microbial data identified alterations of the gut microbiota in COVID-19 patients during the acute and recovery phases. BMC Microbiol. 2022, 22, 274. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Y.; Li, C.; Chang, W.; Zhang, L. The relationship between gut microbiota and COVID-19 progression: New insights into immunopathogenesis and treatment. Front. Immunol. 2023, 14, 1180336. [Google Scholar] [CrossRef]
- Liu, Q.; Su, Q.; Zhang, F.; Tun, H.M.; Mak, J.W.Y.; Lui, G.C.; Ng, S.S.S.; Ching, J.Y.L.; Li, A.; Lu, W.; et al. Multi-kingdom gut microbiota analyses define COVID-19 severity and post-acute COVID-19 syndrome. Nat. Commun. 2022, 13, 6806. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bradley, E.; Zeamer, A.L.; Cincotta, L.; Salive, M.C.; Dutta, P.; Mutaawe, S.; Anya, O.; Meza-Segura, M.; Moormann, A.M.; et al. Inflammation-type dysbiosis of the oral microbiome associates with the duration of COVID-19 symptoms and long COVID. JCI Insight 2021, 6, e152346. [Google Scholar] [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef]
- Almulla, A.F.; Supasitthumrong, T.; Tunvirachaisakul, C.; Algon, A.A.A.; Al-Hakeim, H.K.; Maes, M. The tryptophan catabolite or kynurenine pathway in COVID-19 and critical COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2022, 22, 615. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, M.M.; Sivandzade, F.; Albekairi, T.H.; Alqahtani, F.; Cucullo, L. Neuroinflammation and Its Impact on the Pathogenesis of COVID-19. Front. Med. 2021, 8, 745789. [Google Scholar] [CrossRef]
- Fitero, A.; Bungau, S.G.; Tit, D.M.; Endres, L.; Khan, S.A.; Bungau, A.F.; Romanul, I.; Vesa, C.M.; Radu, A.-F.; Tarce, A.G.; et al. Comorbidities, Associated Diseases, and Risk Assessment in COVID-19—A Systematic Review. Int. J. Clin. Pract. 2022, 2022, 1571826. [Google Scholar] [CrossRef]
- Moghimi, N.; Di Napoli, M.; Biller, J.; Siegler, J.E.; Shekhar, R.; McCullough, L.D.; Harkins, M.S.; Hong, E.; Alaouieh, D.A.; Mansueto, G.; et al. The Neurological Manifestations of Post-Acute Sequelae of SARS-CoV-2 Infection. Curr. Neurol. Neurosci. Rep. 2021, 21, 44. [Google Scholar] [CrossRef] [PubMed]
- Moga, T.D.; Nistor-Cseppento, C.D.; Bungau, S.G.; Tit, D.M.; Sabau, A.M.; Behl, T.; Nechifor, A.C.; Bungau, A.F.; Negrut, N. The Effects of the “Catabolic Crisis” on Patients’ Prolonged Immobility after COVID-19 Infection. Medicina 2022, 58, 828. [Google Scholar] [CrossRef]
- Nistor-Cseppento, C.D.; Moga, T.D.; Bungau, A.F.; Tit, D.M.; Negrut, N.; Pasca, B.; Bochis, C.F.; Ghitea, T.C.; Jurcau, A.; Purza, A.L.; et al. The Contribution of Diet Therapy and Probiotics in the Treatment of Sarcopenia Induced by Prolonged Immobilization Caused by the COVID-19 Pandemic. Nutrients 2022, 14, 4701. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gland | Clinical Manifestation |
|---|---|
| Pituitary gland | Pituitary apoplexy |
| Thyroid gland | Euthyroid sick syndrome Subacute thyroiditis Graves’ disease Postpartum thyroiditis Hashimoto’s disease Silent thyroiditis |
| Pancreas | Acute pancreatitis Necrotizing pancreatitis Impaired glucose tolerance Insulin resistance Diabetes mellitus Severe metabolic complications (diabetic ketoacidosis) Hyperglycemia Abnormalities level of amylase/lipase |
| Adrenal glands | Necrosis Hypocortisolism Cross-reactive antibodies against endogenous adrenocorticotropic hormone |
| Gonads | Hypogonadism Erectile dysfunction Menstrual disturbances |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negrut, N.; Menegas, G.; Kampioti, S.; Bourelou, M.; Kopanyi, F.; Hassan, F.D.; Asowed, A.; Taleouine, F.Z.; Ferician, A.; Marian, P. The Multisystem Impact of Long COVID: A Comprehensive Review. Diagnostics 2024, 14, 244. https://doi.org/10.3390/diagnostics14030244
Negrut N, Menegas G, Kampioti S, Bourelou M, Kopanyi F, Hassan FD, Asowed A, Taleouine FZ, Ferician A, Marian P. The Multisystem Impact of Long COVID: A Comprehensive Review. Diagnostics. 2024; 14(3):244. https://doi.org/10.3390/diagnostics14030244
Chicago/Turabian StyleNegrut, Nicoleta, Georgios Menegas, Sofia Kampioti, Maria Bourelou, Francesca Kopanyi, Faiso Dahir Hassan, Anamaria Asowed, Fatima Zohra Taleouine, Anca Ferician, and Paula Marian. 2024. "The Multisystem Impact of Long COVID: A Comprehensive Review" Diagnostics 14, no. 3: 244. https://doi.org/10.3390/diagnostics14030244
APA StyleNegrut, N., Menegas, G., Kampioti, S., Bourelou, M., Kopanyi, F., Hassan, F. D., Asowed, A., Taleouine, F. Z., Ferician, A., & Marian, P. (2024). The Multisystem Impact of Long COVID: A Comprehensive Review. Diagnostics, 14(3), 244. https://doi.org/10.3390/diagnostics14030244