
The Potential Role of Cell-Death Mechanisms in the Pathogenesis of Familial Mediterranean Fever Attacks: Granzyme A and Beyond

 , , and
, , and
Abstract
1. Introduction
2. Material and Methods
Statistical Analysis
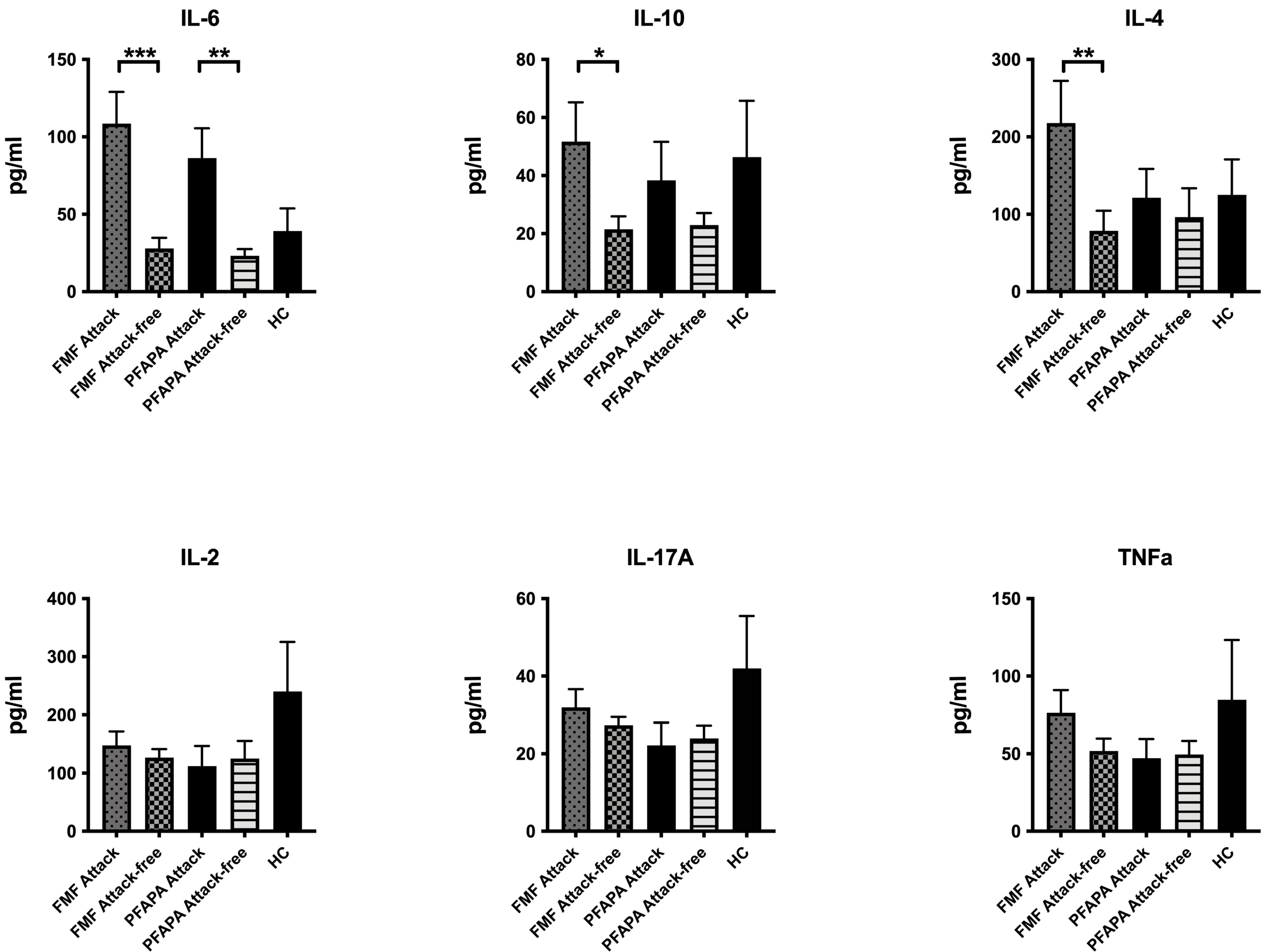
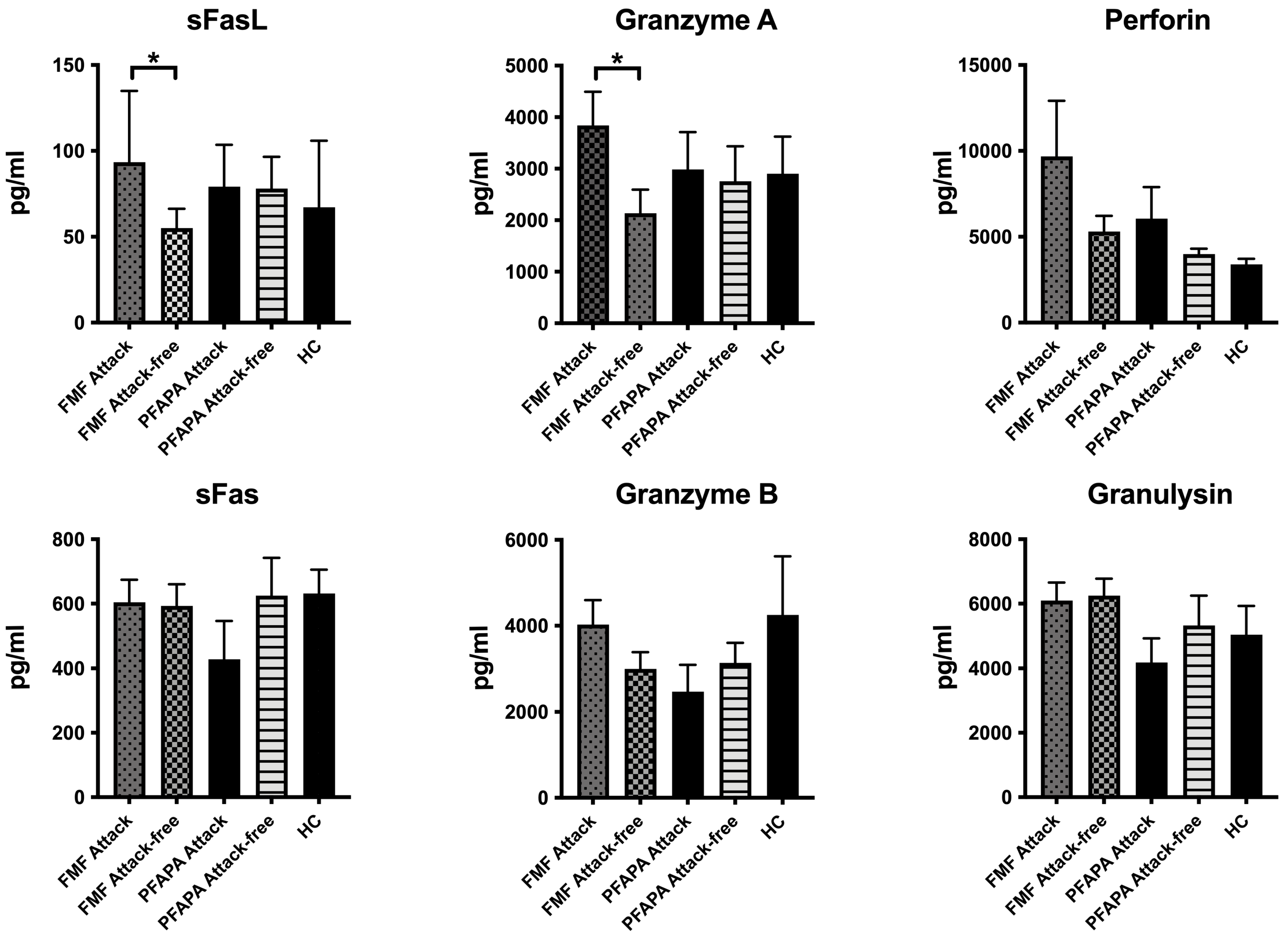
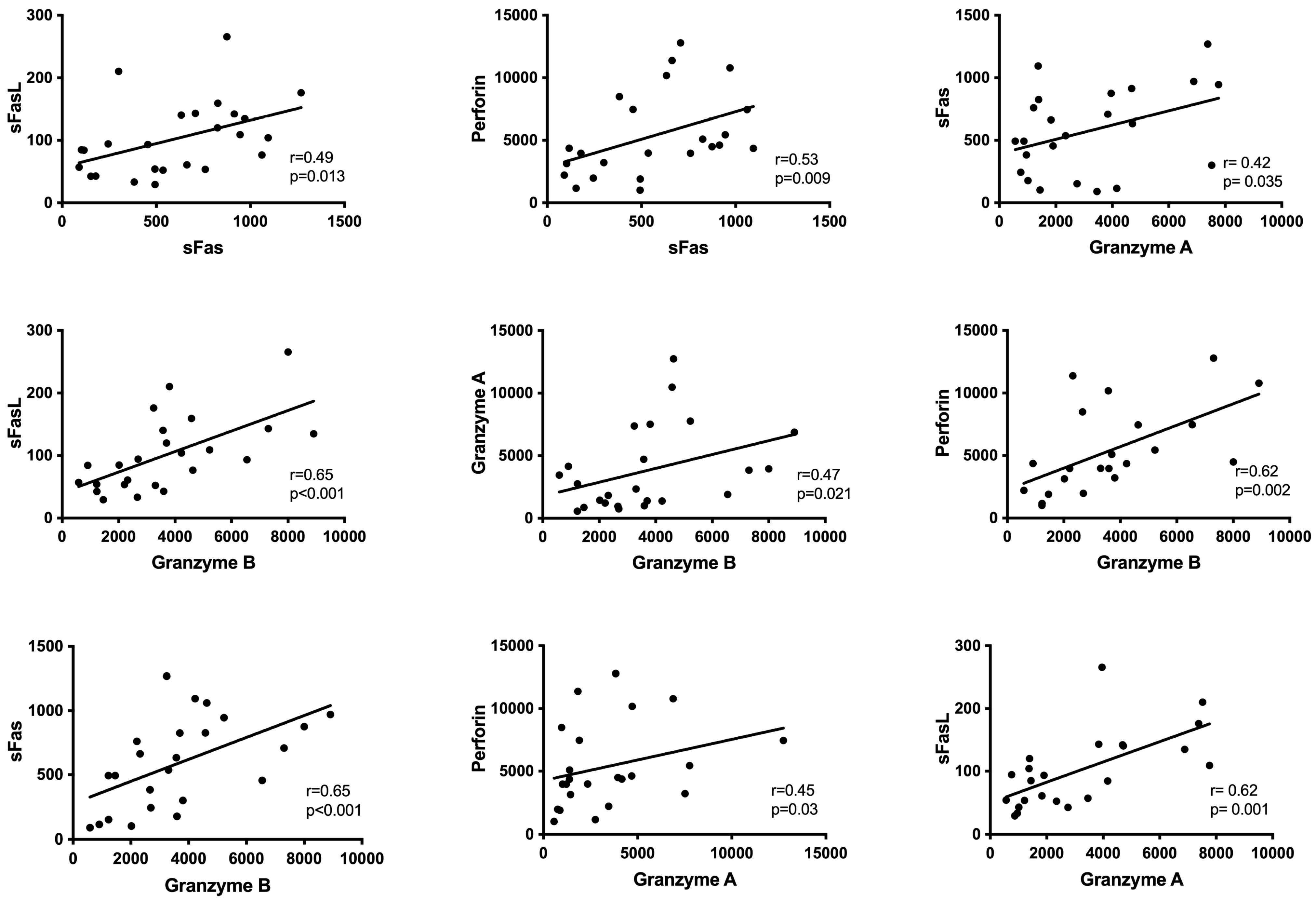
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Touitou, I.; Kone-Paut, I. Autoinflammatory diseases. Best Pract. Res. Clin. Rheumatol. 2008, 22, 811–829. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E.; Ozen, S.; Balci, B.; Duzova, A.; Topaloglu, R.; Besbas, N.; Saatci, U.; Bakkaloglu, A.; Ozguc, M. Mutation frequency of Familial Mediterranean Fever and evidence for a high carrier rate in the Turkish population. Eur. J. Hum. Genet. EJHG 2001, 9, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Karaaslan, Y.; Ozdemir, O.; Saatci, U.; Bakkaloglu, A.; Koroglu, E.; Tezcan, S. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: A field study. J. Rheumatol. 1998, 25, 2445–2449. [Google Scholar] [PubMed]
- The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997, 90, 797–807. [Google Scholar] [CrossRef] [PubMed]
- French, F.M.F.C. A candidate gene for familial Mediterranean fever. Nat. Genet. 1997, 17, 25–31. [Google Scholar] [CrossRef]
- Chae, J.J.; Cho, Y.H.; Lee, G.S.; Cheng, J.; Liu, P.P.; Feigenbaum, L.; Katz, S.I.; Kastner, D.L. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity 2011, 34, 755–768. [Google Scholar] [CrossRef]
- Masters, S.L.; Simon, A.; Aksentijevich, I.; Kastner, D.L. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease. Annu. Rev. Immunol. 2009, 27, 621–668. [Google Scholar] [CrossRef]
- Livneh, A.; Langevitz, P.; Zemer, D.; Zaks, N.; Kees, S.; Lidar, T.; Migdal, A.; Padeh, S.; Pras, M. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997, 40, 1879–1885. [Google Scholar] [CrossRef]
- Yalcinkaya, F.; Ozen, S.; Ozcakar, Z.B.; Aktay, N.; Cakar, N.; Duzova, A.; Kasapcopur, O.; Elhan, A.H.; Doganay, B.; Ekim, M.; et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology 2009, 48, 395–398. [Google Scholar] [CrossRef]
- Gattorno, M.; Hofer, M.; Federici, S.; Vanoni, F.; Bovis, F.; Aksentijevich, I.; Anton, J.; Arostegui, J.I.; Barron, K.; Ben-Cherit, E.; et al. Classification criteria for autoinflammatory recurrent fevers. Ann. Rheum. Dis. 2019, 78, 1025–1032. [Google Scholar] [CrossRef]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Demirkaya, E.; Erer, B.; Ozen, S.; Ben-Chetrit, E. Efficacy and safety of treatments in Familial Mediterranean fever: A systematic review. Rheumatol. Int. 2016, 36, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Livneh, A.; Zemer, D.; Langevitz, P.; Shemer, J.; Sohar, E.; Pras, M. Colchicine in the treatment of AA and AL amyloidosis. Semin. Arthritis Rheum. 1993, 23, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Demirkaya, E.; Erer, B.; Livneh, A.; Ben-Chetrit, E.; Giancane, G.; Ozdogan, H.; Abu, I.; Gattorno, M.; Hawkins, P.N.; et al. EULAR recommendations for the management of familial Mediterranean fever. Ann. Rheum. Dis. 2016, 75, 644–651. [Google Scholar] [CrossRef]
- Sag, E.; Akal, F.; Atalay, E.; Akca, U.K.; Demir, S.; Demirel, D.; Batu, E.D.; Bilginer, Y.; Ozen, S. Anti-IL1 treatment in colchicine-resistant paediatric FMF patients: Real life data from the HELIOS registry. Rheumatology 2020, 59, 3324–3329. [Google Scholar] [CrossRef]
- Tang, L.; Lu, C.; Zheng, G.; Burgering, B.M. Emerging insights on the role of gasdermins in infection and inflammatory diseases. Clin. Transl. Immunol. 2020, 9, e1186. [Google Scholar] [CrossRef]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Pillay, J.; den Braber, I.; Vrisekoop, N.; Kwast, L.M.; de Boer, R.J.; Borghans, J.A.; Tesselaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Pillet, P.; Cochard, M.M.; Berg, S.; Krol, P.; Kone-Paut, I.; Rigante, D.; Hentgen, V.; Anton, J.; Brik, R.; et al. International periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis syndrome cohort: Description of distinct phenotypes in 301 patients. Rheumatology 2014, 53, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G.S.; Edwards, K.M.; Butler, J.; Lawton, A.R. Syndrome of periodic fever, pharyngitis, and aphthous stomatitis. J. Pediatr. 1987, 110, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrin, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef]
- Kanneganti, A.; Malireddi, R.K.S.; Saavedra, P.H.V.; Vande Walle, L.; Van Gorp, H.; Kambara, H.; Tillman, H.; Vogel, P.; Luo, H.R.; Xavier, R.J.; et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J. Exp. Med. 2018, 215, 1519–1529. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- Jorch, S.K.; McNally, A.; Berger, P.; Wolf, J.; Kaiser, K.; Chetrusca Covash, A.; Robeck, S.; Pastau, I.; Fehler, O.; Jauch-Speer, S.L.; et al. Complex regulation of alarmins S100A8/A9 and secretion via gasdermin D pores exacerbates autoinflammation in familial Mediterranean fever. J. Allergy Clin. Immunol. 2023, 152, 230–243. [Google Scholar] [CrossRef]
- Ozen, S.; Uckan, D.; Baskin, E.; Besbas, N.; Okur, H.; Saatci, U.; Bakkaloglu, A. Increased neutrophil apoptosis during attacks of familial Mediterranean fever. Clin. Exp. Rheumatol. 2001, 19, S68–S71. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| FMF Attack | FMF Attack-Free | p | PFAPA Attack | PFAPA Attack-Free | p | |
|---|---|---|---|---|---|---|
| Hemoglobin (g/dL) | 12.6 ± 1.0 | 12.71 ± 1.30 | 0.705 | 10.7 ± 3.6 | 11.9 ± 0.9 | 0.966 |
| WBC (/mm3) | 10,662.4 ± 4937.3 | 8187.90 ± 4625.12 | 0.074 | 11,376.0 ± 4128.7 | 10,237.0 ± 3957.6 | 0.753 |
| Platelet (103/mm3) | 287,040 ± 102,403 | 288,480 ± 75,212 | 0.955 | 309,200 ± 144,024 | 384,600 ± 74,099 | 0.159 |
| Erythrocyte sedimentation rate (mm/hr) | 24.3 ± 15.1 | 10.45 ± 6.12 | <0.001 | 13.9 ± 5.8 | 11.9 ± 10.8 | 0.631 |
| C-reactive protein(mg/dL) | 68.8 ± 69.0 | 6.00 ± 9.73 | <0.001 | 63.4 ± 56.9 | 12.0 ± 17.9 | 0.021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yaglikara, E.; Boluk, O.; Bayindir, Y.; Bilginer, Y.; Tasar, M.A.; Ozen, S.; Sag, E. The Potential Role of Cell-Death Mechanisms in the Pathogenesis of Familial Mediterranean Fever Attacks: Granzyme A and Beyond. Diagnostics 2024, 14, 2031. https://doi.org/10.3390/diagnostics14182031
Yaglikara E, Boluk O, Bayindir Y, Bilginer Y, Tasar MA, Ozen S, Sag E. The Potential Role of Cell-Death Mechanisms in the Pathogenesis of Familial Mediterranean Fever Attacks: Granzyme A and Beyond. Diagnostics. 2024; 14(18):2031. https://doi.org/10.3390/diagnostics14182031
Chicago/Turabian StyleYaglikara, Ece, Oguz Boluk, Yagmur Bayindir, Yelda Bilginer, Medine Aysin Tasar, Seza Ozen, and Erdal Sag. 2024. "The Potential Role of Cell-Death Mechanisms in the Pathogenesis of Familial Mediterranean Fever Attacks: Granzyme A and Beyond" Diagnostics 14, no. 18: 2031. https://doi.org/10.3390/diagnostics14182031
APA StyleYaglikara, E., Boluk, O., Bayindir, Y., Bilginer, Y., Tasar, M. A., Ozen, S., & Sag, E. (2024). The Potential Role of Cell-Death Mechanisms in the Pathogenesis of Familial Mediterranean Fever Attacks: Granzyme A and Beyond. Diagnostics, 14(18), 2031. https://doi.org/10.3390/diagnostics14182031