
Diagnostic Value of Chromosomal Microarray Analysis for Fetal Congenital Heart Defects with Different Cardiac Phenotypes and Extracardiac Abnormalities

Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Classification of Cardiac Phenotypes and ECAs
2.3. Chromosomal Microarray Analysis and Data Analysis
2.4. Statistical Analysis
3. Results
3.1. CMA Results
3.2. Cardiac Phenotype and ECAs in Fetal CHDs
3.2.1. Correlation between CA and ECAs
3.2.2. Correlation between CA and Phenotype of CHDs
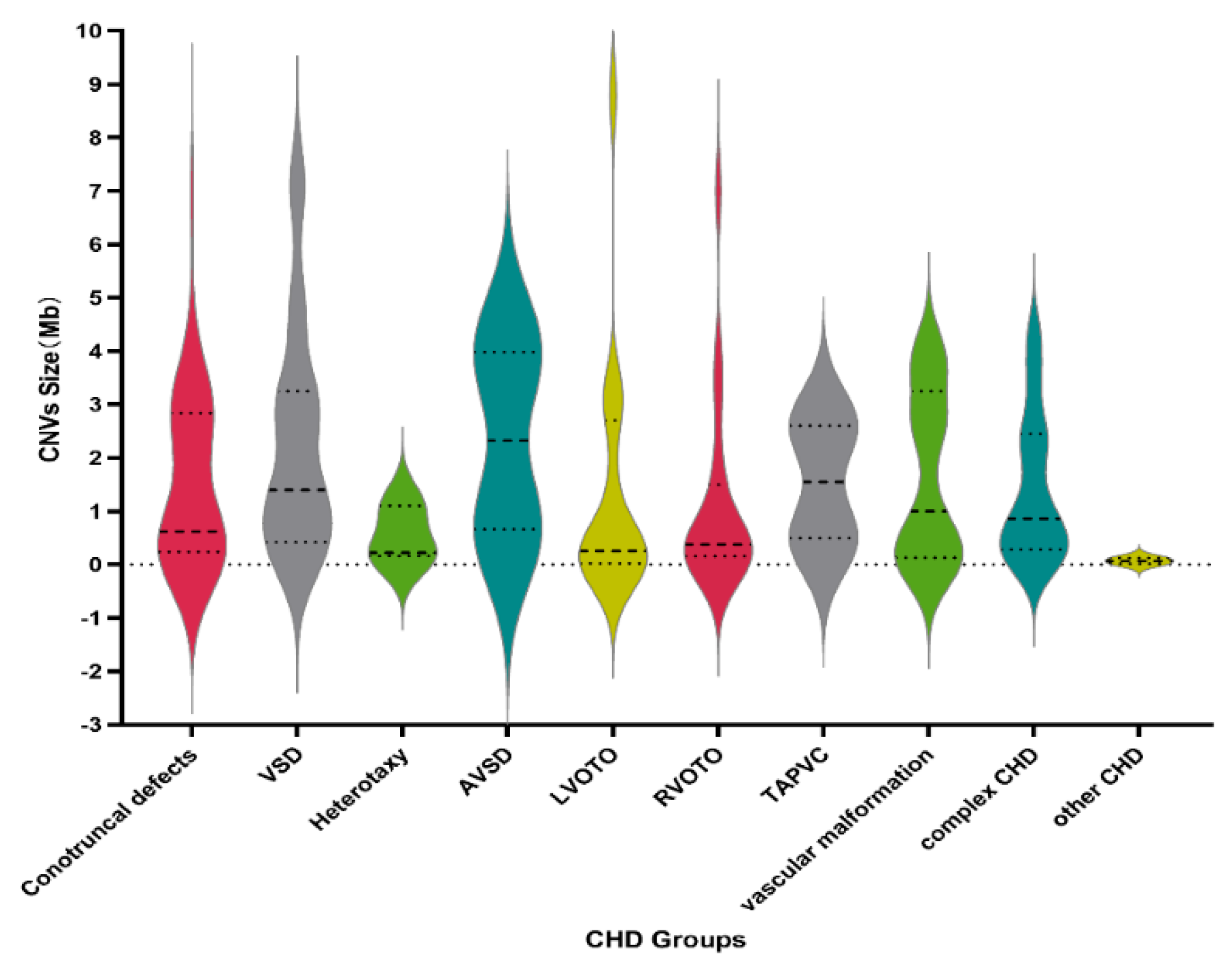
3.3. CNVs Fragment Length in CHD
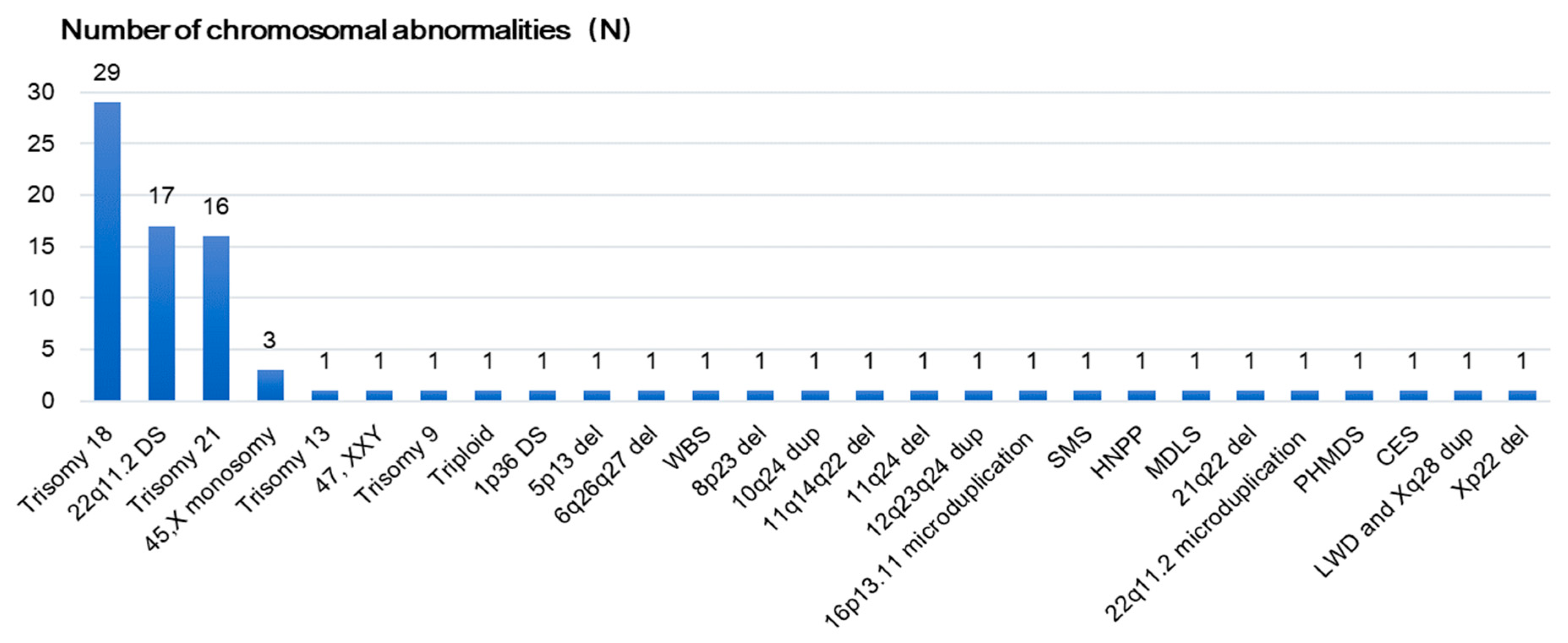
3.4. Detection of CNV Syndrome
3.5. Other Related Pathogenic Genes
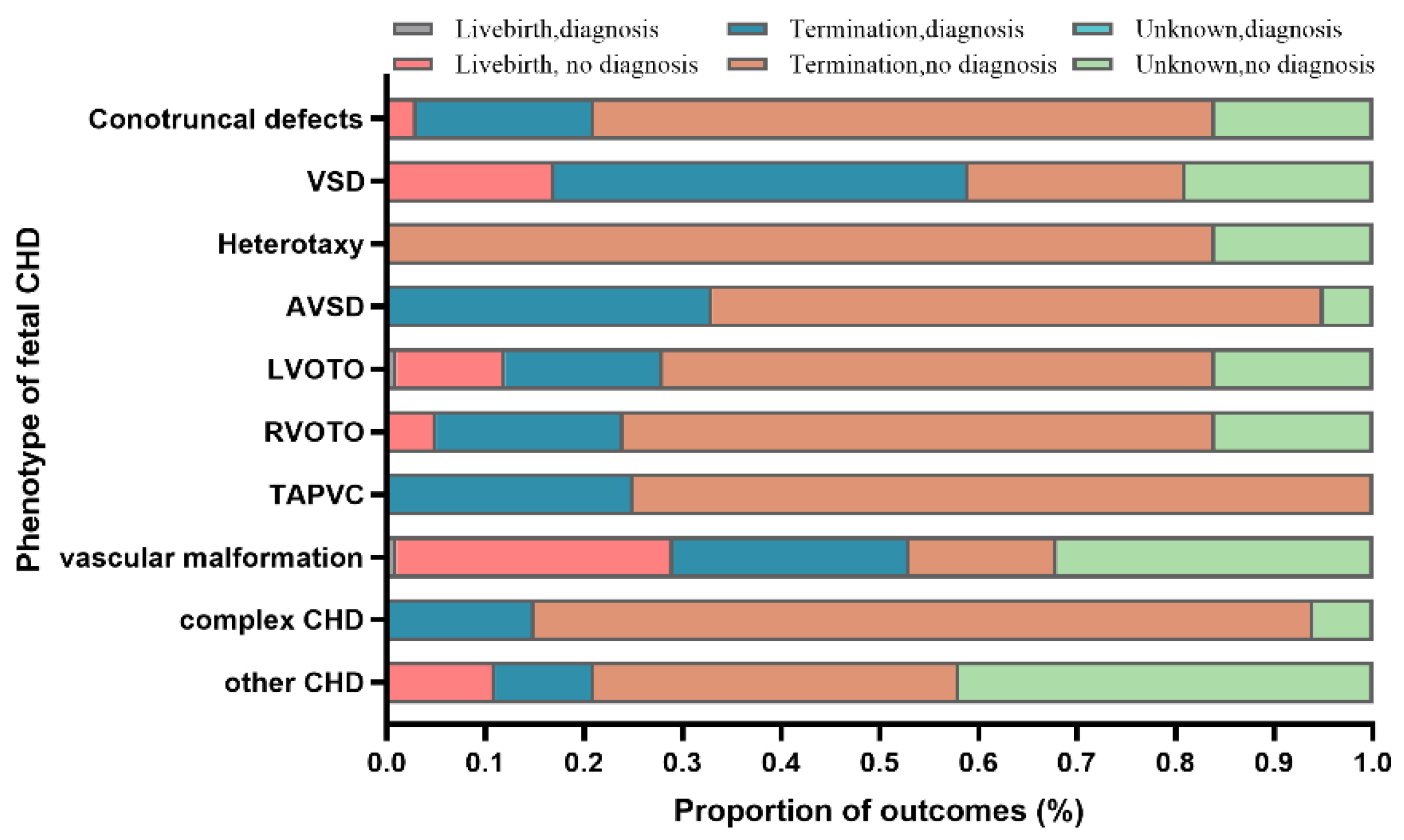
3.6. Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Christianson, A.; Howson, C.P.; Modell, B. March of Dimes: Global Report on Birth Defects, the Hidden Toll of Dying and Disabled Children; White Plains: New York, NY, USA, 2005. [Google Scholar]
- Van Der Bom, T.; Zomer, A.C.; Zwinderman, A.H.; Meijboom, F.J.; Bouma, B.; Mulder, B.J.M. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2011, 8, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Botto, L.D.; Lin, A.E.; Riehle-Colarusso, T.; Malik, S.; Correa, A. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res. Part A Clin. Mol. Teratol. 2007, 79, 714–727. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Y.; Zhang, M.; Lin, N.; An, G.; He, D.; Chen, M.; Chen, L.; Xu, L. Diagnostic accuracy and value of chromosomal microarray analysis for chromosomal abnormalities in prenatal detection: A prospective clinical study. Medicine 2021, 100, e25999. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Tao, J.; Han, X.; Zhao, X.; Liu, C.; Gao, L.; Cheng, W. The clinical use of chromosomal microarray analysis in detection of fetal chromosomal rearrangements: A study from China Mainland. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 212, 44–50. [Google Scholar] [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal Microarray versus Karyotyping for Prenatal Diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef]
- Deng, Q.; Huang, L.; Liu, J.; Fang, F.; Liu, Z.; Zhang, Y.; Li, F.; Liao, C. Prenatal diagnosis of submicroscopic chromosomal aberrations in fetuses with congenital cystic adenomatoid malformation by chromosomal microarray analysis. J. Matern.-Fetal Neonatal Med. 2021, 34, 2623–2629. [Google Scholar] [CrossRef]
- Dovjak, G.O.; Zalewski, T.; Seidl-Mlczoch, E.; Ulm, P.A.; Berger-Kulemann, V.; Weber, M.; Prayer, D.; Kasprian, G.J.; Ulm, B. Abnormal Extracardiac Development in Fetuses with Congenital Heart Disease. J. Am. Coll. Cardiol. 2021, 78, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Hu, A.; Dyhamenahali, U.; Chitayat, D.; Winsor, E.J.T.; Ryan, G.; Smallhorn, J.; Barrett, J.; Yoo, S.-J.; Hornberger, L.K. Extracardiac lesions and chromosomal abnormalities associated with major fetal heart defects: Comparison of intrauterine, postnatal and postmortem diagnoses. Ultrasound Obstet. Gynecol. 2009, 33, 552–559. [Google Scholar] [CrossRef]
- Bensemlali, M.; Bajolle, F.; Ladouceur, M.; Fermont, L.; Lévy, M.; Le Bidois, J.; Salomon, L.J.; Bonnet, D. Associated genetic syndromes and extracardiac malformations strongly influence outcomes of fetuses with congenital heart diseases. Arch. Cardiovasc. Dis. 2016, 109, 330–336. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, L.; Liang, D.; Meng, L.; Wu, Y.; Qiao, F.; Ji, X.; Luo, C.; Zhang, J.; Xu, T.; et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: A prospective cohort study. Am. J. Obstet. Gynecol. 2018, 218, 244.e1–244.e17. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Wang, Y.; Zhang, C.; Zhou, R.; Wu, Y.; Wang, C.; Meng, L.; Mao, P.; Cheng, Q.; Luo, C.; et al. Comprehensive evaluation of genetic variants using chromosomal microarray analysis and exome sequencing in fetuses with congenital heart defect. Ultrasound Obstet. Gynecol. 2021, 58, 377–387. [Google Scholar] [CrossRef]
- Digilio, M.C.; Marino, B. What Is New in Genetics of Congenital Heart Defects? Front. Pediatr. 2016, 4, 120. [Google Scholar] [CrossRef]
- Moyano, D.; Huggon, I.C.; Allan, L.D. Fetal echocardiography in trisomy 18. Arch. Dis. Child. Fetal Neonatal Ed. 2005, 90, F520–F522. [Google Scholar] [CrossRef]
- Postema, P.G.; Rammeloo, L.A.; van Litsenburg, R.; Rothuis, E.G.; Hruda, J. Left superior vena cava in pediatric cardiology associated with extra-cardiac anomalies. Int. J. Cardiol. 2008, 123, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Sun, H.; Fu, Y.; Hao, X.; Sun, L.; Zhang, Y.; Han, J.; Gu, X.; Liu, X.; Guo, Y.; et al. Genetic and Clinical Features of Heterotaxy in a Prenatal Cohort. Front. Genet. 2022, 13, 818241. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, L.G.; Rosenfeld, J.A.; Dabell, M.P.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Fisher, A.J. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat. Diagn. 2012, 32, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Li, R.; Fu, F.; Xie, G.; Zhang, Y.; Pan, M.; Li, J.; Li, D. Prenatal diagnosis of congenital heart defect by genome-wide high-resolution SNP array. Prenat. Diagn. 2014, 34, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wu, Q.; Zhang, L.; Wang, X.; Dan, S.; Deng, D.; Sun, L.; Yao, L.; Ma, Y.; Wang, L. Detection of submicroscopic chromosomal aberrations by array-based comparative genomic hybridization in fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 2014, 43, 404–412. [Google Scholar] [CrossRef]
- Savory, K.; Manivannan, S.; Zaben, M.; Uzun, O.; Syed, Y.A. Impact of copy number variation on human neurocognitive deficits and congenital heart defects: A systematic review. Neurosci. Biobehav. Rev. 2020, 108, 83–93. [Google Scholar] [CrossRef]
- Shaw, C.J.; Lupski, J.R. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum. Genet. 2005, 116, 1–7. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002, 18, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef]
- Hou, H.-T.; Chen, H.-X.; Wang, X.-L.; Yuan, C.; Yang, Q.; Liu, Z.-G.; He, G.-W. Correction: Genetic characterisation of 22q11.2 variations and prevalence in patients with congenital heart disease. Arch. Dis. Child. 2020, 105, e7. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Wang, S. Detection of chromosomal abnormalities and the 22q11 microdeletion in fetuses with congenital heart defects. Mol. Med. Rep. 2014, 10, 2465–2470. [Google Scholar] [CrossRef]
- Li, Z.; Huang, J.; Liang, B.; Zeng, D.; Luo, S.; Yan, T.; Liao, F.; Huang, J.; Li, J.; Cai, R.; et al. Copy number variations in the GATA4, NKX2-5, TBX5, BMP4 CRELD1, and 22q11.2 gene regions in Chinese children with sporadic congenital heart disease. J. Clin. Lab. Anal. 2019, 33, e22660. [Google Scholar] [CrossRef]
- Ben-Shachar, S.; Ou, Z.; Shaw, C.A.; Belmont, J.W.; Patel, M.S.; Hummel, M.; Amato, S.; Tartaglia, N.; Berg, J.; Sutton, V.R.; et al. 22q11.2 Distal Deletion: A Recurrent Genomic Disorder Distinct from DiGeorge Syndrome and Velocardiofacial Syndrome. Am. J. Hum. Genet. 2008, 82, 214–221. [Google Scholar] [CrossRef]
- Mlynarski, E.E.; Sheridan, M.B.; Xie, M.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Gai, X.; Chow, E.W.; Vorstman, J.; Swillen, A.; et al. Copy-Number Variation of the Glucose Transporter Gene SLC2A3 and Congenital Heart Defects in the 22q11.2 Deletion Syndrome. Am. J. Hum. Genet. 2015, 96, 753–764. [Google Scholar] [CrossRef]
- Zhao, Y.; Edington, S.; Fleenor, J.; Sinkovskaya, E.; Porche, L.; Abuhamad, A. Fetal cardiac axis in tetralogy of Fallot: Associations with prenatal findings, genetic anomalies and postnatal outcome. Ultrasound Obstet. Gynecol. 2017, 50, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R. Other genomic disorders and congenital heart disease. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 107–115. [Google Scholar] [CrossRef]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gécz, J.; et al. Duplication of the MECP2 Region Is a Frequent Cause of Severe Mental Retardation and Progressive Neurological Symptoms in Males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef]
- Matsuoka, R.; Yoshida, M.C.; Furutani, Y.; Imamura, S.-I.; Kanda, N.; Yanagisawa, M.; Masaki, T.; Takao, A. Human smooth muscle myosin heavy chain gene mapped to chromosomal region 16q12. Am. J. Med. Genet. 1993, 46, 61–67. [Google Scholar] [CrossRef]
- Morano, I.; Chai, G.-X.; Baltas, L.G.; Lamounier-Zepter, V.; Lutsch, G.; Kott, M.; Haase, H.; Bader, M. Smooth-muscle contraction without smooth-muscle myosin. Nature 2000, 2, 371–375. [Google Scholar] [CrossRef]
- Hart, A.; Melet, F.; Grossfeld, P.; Chien, K.; Jones, C.; Tunnacliffe, A.; Favier, R.; Bernstein, A. Fli-1 Is Required for Murine Vascular and Megakaryocytic Development and Is Hemizygously Deleted in Patients with Thrombocytopenia. Immunity 2000, 13, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, E.; Wang, T.-J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef]
- D’Alessandro, L.C.A.; Al Turki, S.; Manickaraj, A.K.; Manase, D.; Mulder, B.J.M.; Bergin, L.; Rosenberg, H.C.; Mondal, T.; Gordon, E.; Lougheed, J.; et al. Exome sequencing identifies rare variants in multiple genes in atrioventricular septal defect. Genet. Med. 2016, 18, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Zirnsak, B.; Segebrecht, L.; Schubach, M.; Charles, P.; Alderman, E.; Brown, K.; Cadieux-Dion, M.; Cartwright, T.; Chen, Y.; Costin, C.; et al. Haploinsufficiency of the Notch Ligand DLL1 Causes Variable Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.M., Jr.; Kramer, N.; Bejjani, B.A.; Thiel, C.T.; Carta, C.; Neri, G.; Tartaglia, M.; Zenker, M. Genomic duplication of PTPN11 is an uncommon cause of Noonan syndrome. Am. J. Med. Genet. Part A 2009, 149a, 2122–2128. [Google Scholar] [CrossRef]
- Moskowitz, I.P.; Kim, J.B.; Moore, M.L.; Wolf, C.M.; Peterson, M.A.; Shendure, J.; Nobrega, M.A.; Yokota, Y.; Berul, C.; Izumo, S.; et al. A Molecular Pathway Including Id2, Tbx5, and Nkx2-5 Required for Cardiac Conduction System Development. Cell 2007, 129, 1365–1376. [Google Scholar] [CrossRef]
- Hiroi, Y.; Kudoh, S.; Monzen, K.; Ikeda, Y.; Yazaki, Y.; Nagai, R.; Komuro, I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat. Genet. 2001, 28, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Morello, M.L.; Beidelman, M.; Saha, P.; Ingelsson, E.; Shaw, G.; Lui, G.; Priest, J. Neurocognitive deficits in adult congenital heart disease: Does coronary artery disease add insult to injury? J. Am. Coll. Cardiol. 2019, 73, 566. [Google Scholar] [CrossRef]
- Morton, P.D.; Ishibashi, N.; Jonas, R.A. Neurodevelopmental Abnormalities and Congenital Heart Disease: Insights Into Altered Brain Maturation. Circ. Res. 2017, 120, 960–977. [Google Scholar] [CrossRef]
- Donofrio, M.T.; Moon-Grady, A.J.; Hornberger, L.K.; Copel, J.A.; Sklansky, M.S.; Abuhamad, A.; Cuneo, B.F.; Huhta, J.C.; Jonas, R.A.; Krishnan, A.; et al. Diagnosis and Treatment of Fetal Cardiac Disease: A scientific statement from the American Heart Association. Circulation 2014, 129, 2183–2242. [Google Scholar] [CrossRef]
- McQuillen, P.S.; Goff, D.A.; Licht, D.J. Effects of congenital heart disease on brain development. Prog. Pediatr. Cardiol. 2010, 29, 79–85. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Groups | Total | Isolated CHDs | ECAs | Total vs. Isolated vs. ECAs | Isolated vs. ECAs | |||
|---|---|---|---|---|---|---|---|---|
| n | CA(n(%)) | n | CA(n(%)) | n | CA(n(%)) | p | p | |
| Conotruncal defects | 138 | 25(18.1) | 104(34.0) | 13(12.5) | 34(28.1) | 12(35.3) | 0.011 | 0.003 |
| Septal defect (VSD) | 60 | 25(41.7) | 28(9.2) | 11(39.3) | 32(26.4) | 14(43.8) | 0.941 | |
| LVOTO | 47 | 8(17.0) | 35(11.4) | 5(14.3) | 12(9.9) | 3(25.0) | 0.711 | |
| RVOTO | 42 | 8(19.0) | 31(10.1) | 4(12.9) | 11(9.1) | 4(36.4) | 0.267 | |
| AVSD | 39 | 13(33.3) | 23(7.5) | 6(26.1) | 16(13.2) | 7(43.8) | 0.516 | |
| TAPVC | 4 | 1(25.0) | 3(1.0) | 0(0) | 1(0.8) | 1(100) | 0.669 | |
| vascular abnormality | 22 | 6(27.3) | 19(6.2) | 5(26.3) | 3(2.5) | 1(33.3) | 0.990 | |
| Heterotaxy (complex CHD) | 25 | 0(0) | 23(7.5) | 0(0) | 2(1.7) | 0(0) | ||
| Complex CHD (Multiple, Single ventricle) | 33 | 5(15.2) | 28(9.2) | 4(14.3) | 5(4.1) | 1(20.0) | 0.896 | |
| Other* | 17 | 2(11.8) | 12(3.9) | 1(8.3) | 5(4.1) | 1(20.0) | 0.765 | |
| Total | 427 | 93(21.8) | 306(100.0) | 49(16.0) | 121(100.0) | 44(36.4) | 0.000 | 0.000 |
| Category | n | CA(n(%)) | NCA | pCNVs |
|---|---|---|---|---|
| Craniofacial abnormality | ||||
| Yes | 24(19.8) | 10(41.7) | 8 | 2 |
| No | 97(80.2) | 34(35.1) | 19 | 15 |
| Neurologic abnormality | ||||
| Yes | 34(28.1) | 10(29.4) | 4 | 6 |
| No | 87(71.9) | 34(39.1) | 23 | 11 |
| Skeletal abnormality | ||||
| Yes | 39(32.2) | 19(48.7) | 15 | 4 |
| No | 82(67.8) | 25(30.5) | 12 | 13 |
| Digestive abnormality | ||||
| Yes | 18(14.9) | 5(27.8) | 4 | 1 |
| No | 103(85.1) | 39(37.9) | 23 | 16 |
| Urogenital abnormality | ||||
| Yes | 31(25.6) | 10(32.3) | 6 | 4 |
| No | 90(74.4) | 34(37.7) | 21 | 13 |
| Thoracic mass lesion | ||||
| Yes | 14(11.6) | 6(42.9) | 4 | 2 |
| No | 107(88.4) | 38(35.5) | 23 | 15 |
| Thoracic & abdominal wall abnormality | ||||
| Yes | 7(5.8) | 4(57.2) | 4 | 0 |
| No | 114(94.2) | 40(35.1) | 23 | 17 |
| Thymic abnormality | ||||
| Yes | 5(4.1) | 5(100) | 0 | 5 |
| No | 116(95.9) | 39(33.6) | 27 | 12 |
| Types of ECAs | ||||
| One | 79(65.3) | 24(30.4) | 13 | 11 |
| Two or more | 42(34.7) | 20(47.6) | 14 | 6 |
| Total | 121 | 44(36.4) | 27 | 17 |
| Groups and Subgroups | n | CA(n(%)) | NCA(n(%)) | pCNVs(n(%)) | T21 | T18 | T13 | 45, X | Othe NCA | 22q11.2 DS | Other CNV Syndrome | Other pCNVs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conotruncal defects | 138 | 25(18.7) | 11(8.0) | 14(10.1) | 1 | 8 | 0 | 0 | 2 | 10 | 2 | 2 |
| TOF | 68 | 12(17.6) | 3(4.4) | 9(13.2) | 0 | 3 | 0 | 0 | 0 | 7 | 1 | 1 |
| CAT | 16 | 3(18.8) | 1(6.3) | 2(12.5) | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 |
| d-TGA | 16 | 1(6.3) | 0(0) | 1(6.3) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| DORV | 35 | 8(22.9) | 7(20.0) | 1(2.9) | 1 | 4 | 0 | 0 | 2 | 1 | 0 | 0 |
| IAA, type B | 3 | 1(33.3) | 0(0) | 1(33.3) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Septal defect (VSD) | 60 | 25(41.7) | 19(31.7) | 6(10.0) | 4 | 14 | 1 | 0 | 0 | 0 | 5 | 1 |
| LVOTO | 47 | 8(17.0) | 2(4.3) | 6(12.7) | 0 | 0 | 0 | 2 | 0 | 3 | 1 | 2 |
| Aortic stenosis | 14 | 2(14.3) | 0(0) | 1(14.3) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Coarctation of aorta (CoA) | 9 | 4(44.4) | 2(22.2) | 2(22.2) | 0 | 0 | 0 | 2 | 0 | 1 | 1 | 0 |
| HLHS | 20 | 1(5.0) | 0(0) | 1(5.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Mitral valve dysplasia | 2 | 0(0) | 0(0) | 0(0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| IAA, type A | 2 | 1(50.0) | 0(0) | 1(50.0) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| RVOTO | 42 | 8(19.0) | 4(9.5) | 4(9.5) | 3 | 1 | 0 | 0 | 0 | 2 | 0 | 2 |
| HRHS | 9 | 1(11.1) | 0(0) | 1(11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Ebstein’s anomaly, Tricuspid valve dysplasia | 8 | 2(25.0) | 2(25.0) | 0(0) | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PA-IVS | 8 | 0(0) | 0(0) | 0(0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PS | 17 | 5(29.4) | 2(11.8) | 3(17.6) | 1 | 1 | 0 | 0 | 0 | 2 | 0 | 1 |
| AVSD | 39 | 13(33.3) | 13(33.3) | 0(0) | 5 | 5 | 1 | 1 | 1 | 0 | 0 | 0 |
| TAPVC | 4 | 1(25.0) | 0(0) | 1(25.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| vascular abnormality | 22 | 6(27.3) | 2(9.0) | 4(18.2) | 2 | 0 | 0 | 0 | 0 | 3 | 0 | 1 |
| RAA | 14 | 4(28.6) | 0(0) | 4(28.6) | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 1 |
| Double aortic arch | 2 | 0(0) | 0(0) | 0(0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| left superior vena cava | 6 | 2(33.3) | 2(33.3) | 0(0) | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Heterotaxy (complex CHD) | 25 | 0(0) | 0(0) | 0(0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Complex CHD (Multiple, Single ventricle) | 33 | 5(15.2) | 2(6.1) | 3(9.1) | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 |
| other | 17 | 2(11.8) | 2(11.8) | 0(0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rhabdomyoma | 8 | 0(0) | 0(0) | 0(0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hydropericardium | 5 | 2(40.0) | 2(40.0) | 0(0) | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Abnormal heart rhythm And cardiac function | 4 | 0(0) | 0(0) | 0(0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 427 | 93(21.8) | 55(12.9) | 38(8.9) | 17 | 29 | 3 | 3 | 3 | 19 | 10 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Wang, J.; Pei, Y.; Han, J.; Xiong, X.; Yan, Y.; Zhang, J.; Liu, Y.; Su, F.; Xu, J.; et al. Diagnostic Value of Chromosomal Microarray Analysis for Fetal Congenital Heart Defects with Different Cardiac Phenotypes and Extracardiac Abnormalities. Diagnostics 2023, 13, 1493. https://doi.org/10.3390/diagnostics13081493
Zhang S, Wang J, Pei Y, Han J, Xiong X, Yan Y, Zhang J, Liu Y, Su F, Xu J, et al. Diagnostic Value of Chromosomal Microarray Analysis for Fetal Congenital Heart Defects with Different Cardiac Phenotypes and Extracardiac Abnormalities. Diagnostics. 2023; 13(8):1493. https://doi.org/10.3390/diagnostics13081493
Chicago/Turabian StyleZhang, Simin, Jingjing Wang, Yan Pei, Jijing Han, Xiaowei Xiong, Yani Yan, Juan Zhang, Yan Liu, Fangfei Su, Jinyu Xu, and et al. 2023. "Diagnostic Value of Chromosomal Microarray Analysis for Fetal Congenital Heart Defects with Different Cardiac Phenotypes and Extracardiac Abnormalities" Diagnostics 13, no. 8: 1493. https://doi.org/10.3390/diagnostics13081493
APA StyleZhang, S., Wang, J., Pei, Y., Han, J., Xiong, X., Yan, Y., Zhang, J., Liu, Y., Su, F., Xu, J., & Wu, Q. (2023). Diagnostic Value of Chromosomal Microarray Analysis for Fetal Congenital Heart Defects with Different Cardiac Phenotypes and Extracardiac Abnormalities. Diagnostics, 13(8), 1493. https://doi.org/10.3390/diagnostics13081493