
Cancer Diagnosis through Contour Visualization of Gene Expression Leveraging Deep Learning Techniques

 , , , and
, , , and
Abstract
:1. Introduction
- To leverage the power of EBS and JSD as an information-theoretic measure to quantify distributional differences between cancerous and non-cancerous samples based on preprocessed data. By integrating JSD into the analysis, this research aims to gain deeper insights into gene expression patterns, enabling the identification of critical genomic signatures associated with cancer.
- To harness the capabilities of deep learning models for automatic feature extraction and pattern recognition from gene expression data. By employing deep learning, this research seeks to uncover complex molecular relationships and identify crucial features that contribute to accurate cancer detection.
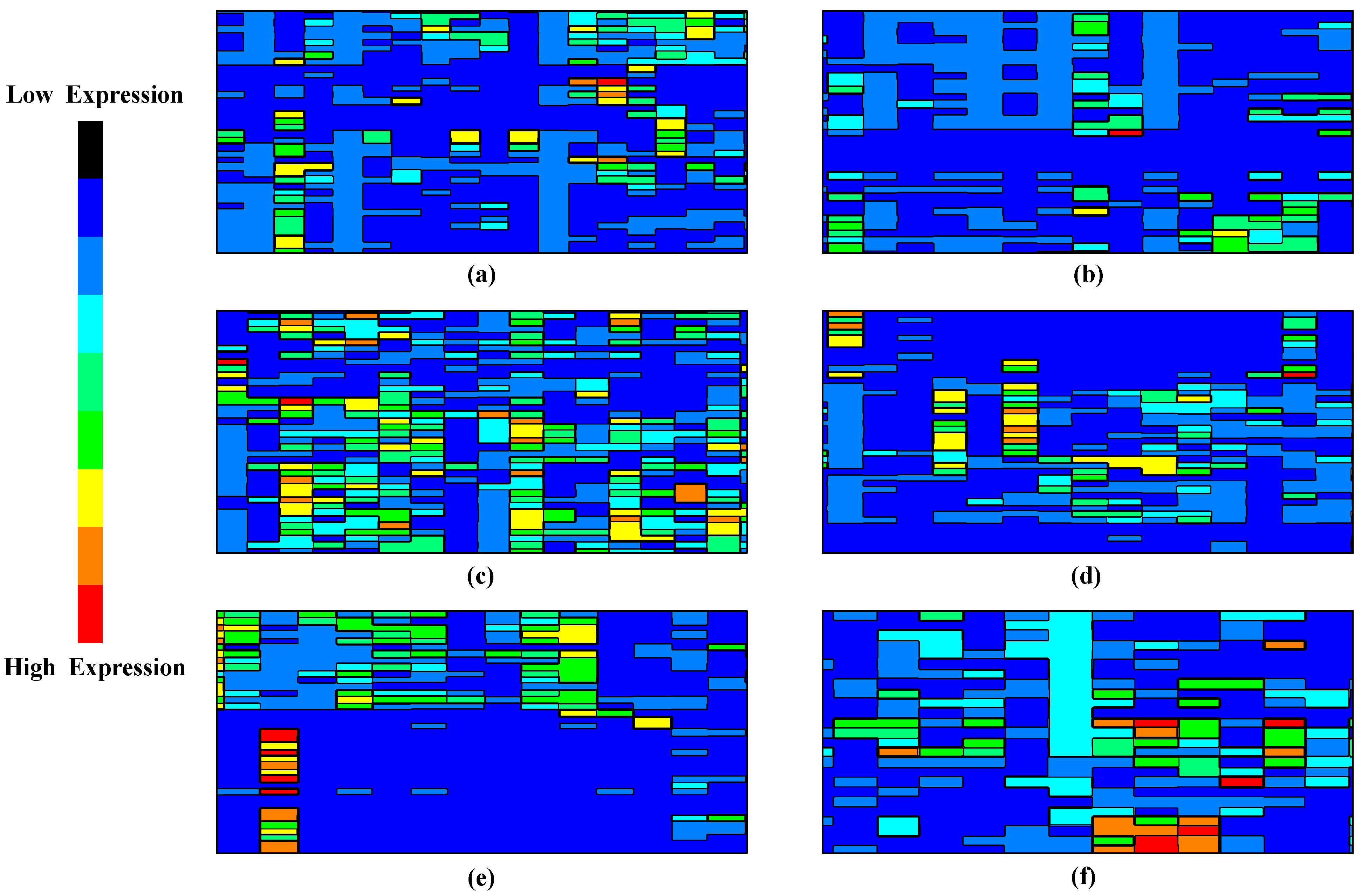
- To utilize contour mathematics for visual interpretation of the deep learning model’s decision boundaries and regions in the high-dimensional feature space. This novel visualization approach enhances the interpretability of the model, facilitating a deeper understanding of the complex interactions between genes and their relevance in cancer detection.
2. Related Work
3. Dataset
4. Methodology
4.1. Data Preprocessing
| Algorithm 1. EBH algorithm. |
| Input: Gene expression data matrix: Output: DI |
| //Data Preparation: 1: split(D) //Bs: biological signal matrix and //Be: Batch-specific effect matrix //Model Fitting: 2: ∀Do ; End Do //Harmonization: 3: ∀ //j = 2, 3, …, k End ∀ 4: ∀Do ; End Do //Batch Effect Correction and Harmonization: 5: 6: //Integration () 7: //integrated dataset |
4.2. Jensen–Shannon Divergence (JSD)
4.3. Intelligent Computation
5. Performance Evaluation
5.1. Empirical Layout
5.2. Outcome Analysis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abbas, Z.; ur Rehman, M.; Tayara, H.; Zou, Q.; Chong, K.T. XGBoost framework with feature selection for the prediction of RNA N5-methylcytosine sites. Mol. Ther. 2023, 31, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Dong, J.; Liu, Y.; Qian, Y.; Zhang, G.; Zhou, W.; Zhao, A.; Ji, G.; Xu, H. New insights into natural products that target the gut microbiota: Effects on the prevention and treatment of colorectal cancer. Front. Pharmacol. 2022, 13, 964793. [Google Scholar] [CrossRef] [PubMed]
- de Guia, J.M.; Devaraj, M.; Leung, C.K. DeepGx. In Proceedings of the 2019 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining, Vancouver, BC, Canada, 27–30 August 2019. [Google Scholar] [CrossRef]
- Gunavathi, C.; Sivasubramanian, K.; Keerthika, P.; Paramasivam, C. A review on convolutional neural network based deep learning methods in gene expression data for disease diagnosis. Mater. Today Proc. 2021, 45, 2282–2285. [Google Scholar] [CrossRef]
- Chen, X.; Liao, Y.; Long, D.; Yu, T.; Shen, F.; Lin, X. The Cdc2/Cdk1 inhibitor, purvalanol A, enhances the cytotoxic effects of taxol through stathmin in non-small cell lung cancer cells in vitro. Int. J. Mol. Med. 2018, 40, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Haznedar, B.; Arslan, M.T.; Kalinli, A. Microarray Gene Expression Cancer Data. Mendeley Data 2017, 2, V4. [Google Scholar] [CrossRef]
- Kazuyuki, H.; Saito, D.; Shouno, H. Analysis of Function of Rectified Linear Unit Used in Deep Learning. In Proceedings of the 2015 International Joint Conference on Neural Networks (IJCNN), Killarney, Ireland, 12–17 July 2015. [Google Scholar] [CrossRef]
- Peng, P.; Luan, Y.; Sun, P.; Wang, L.; Zeng, X.; Wang, Y.; Chen, Y. Prognostic Factors in Stage IV Colorectal Cancer Patients With Resection of Liver and/or Pulmonary Metastases: A Population-Based Cohort Study. Front. Oncol. 2022, 12, 850937. [Google Scholar] [CrossRef]
- Kegerreis, B.; Catalina, M.D.; Bachali, P.; Geraci, N.S.; Labonte, A.C.; Zeng, C.; Stearrett, N.; Crandall, K.A.; Lipsky, P.E.; Grammer, A.C. Machine learning approaches to predict lupus disease activity from gene expression data. Sci. Rep. 2019, 9, 9617. [Google Scholar] [CrossRef]
- Khalifa, N.E.M.; Taha, M.H.N.; Ezzat Ali, D.; Slowik, A.; Hassanien, A.E. Artificial Intelligence Technique for Gene Expression by Tumor RNA-Seq Data: A Novel Optimized Deep Learning Approach. IEEE Access 2020, 8, 22874–22883. [Google Scholar] [CrossRef]
- Li, S.; Yu, J.; Li, M.; Liu, L.; Zhang, X.; Yuan, X. A Framework for Multiclass Contour Visualization. IEEE Trans. Vis. Comput. Graph. 2022, 29, 353–362. [Google Scholar] [CrossRef]
- López-García, G.; Jerez, J.M.; Franco, L.; Veredas, F.J. Transfer learning with convolutional neural networks for cancer survival prediction using gene-expression data. PLoS ONE 2020, 15, e0230536. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.; Cheng, N.; Xiao, J. Dropout Regularization for Self-Supervised Learning of Transformer Encoder Speech Representation. Interspeech 2021, 2021, 1169–1173. [Google Scholar] [CrossRef]
- Maghooli, K.; Moteghaed, N.; Garshasbi, M. Improving Classification of Cancer and Mining Biomarkers from Gene Expression Profiles Using Hybrid Optimization Algorithms and Fuzzy Support Vector Machine. J. Med. Signals Sens. 2018, 8, 1. [Google Scholar] [CrossRef]
- Mahesh, T.R.; Vinoth Kumar, V.; Vivek, V.; Karthick Raghunath, K.M.; Sindhu Madhuri, G. Early predictive model for breast cancer classification using blended ensemble learning. Int. J. Syst. Assur. Eng. Manag. 2022. [Google Scholar] [CrossRef]
- Majumder, S.; Yogita Pal, V.; Yadav, A.; Chakrabarty, A. Performance Analysis of Deep Learning Models for Binary Classification of Cancer Gene Expression Data. J. Healthc. Eng. 2022, 2022, 1122536. [Google Scholar] [CrossRef] [PubMed]
- Maritz, J.S.; Lwin, T. Introduction to Bayes and empirical Bayes methods. In Empirical Bayes Methods; Routledge: London, UK, 2018; pp. 1–26. [Google Scholar] [CrossRef]
- Mir, B.A.; Rehman, M.U.; Tayara, H.; Chong, K.T. Improving Enhancer Identification with a Multi-Classifier Stacked Ensemble Model. J. Mol. Biol. 2023, 435, 168314. [Google Scholar] [CrossRef]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine Learning and Integrative Analysis of Biomedical Big Data. Genes 2019, 10, 87. [Google Scholar] [CrossRef]
- Xie, X.; Wang, X.; Liang, Y.; Yang, J.; Wu, Y.; Li, L.; Shi, X. Evaluating Cancer-Related Biomarkers Based on Pathological Images: A Systematic Review. Front. Oncol. 2021, 11, 763527. [Google Scholar] [CrossRef]
- Nielsen, F. On a Generalization of the Jensen–Shannon Divergence and the Jensen–Shannon Centroid. Entropy 2020, 22, 221. [Google Scholar] [CrossRef]
- Nitta, Y.; Borders, M.; Ludwig, S.A. Analysis of Gene Expression Cancer Data Set: Classification of TCGA Pan-cancer HiSeq Data. In Proceedings of the 2021 IEEE International Conference on Big Data (Big Data), Orlando, FL, USA, 15–18 December 2021. [Google Scholar] [CrossRef]
- Pulumati, A.; Pulumati, A.; Dwarakanath, B.S.; Verma, A.; Papineni, R.V.L. Technological advancements in cancer diagnostics: Improvements and limitations. Cancer Rep. 2023, 6, e1764. [Google Scholar] [CrossRef]
- Rehman, M.U.; Tayara, H.; Chong, K.T. DL-m6A: Identification of N6-Methyladenosine Sites in Mammals Using Deep Learning Based on Different Encoding Schemes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 904–911. [Google Scholar] [CrossRef]
- Rehman, M.U.; Tayara, H.; Zou, Q.; Chong, K.T. i6mA-Caps: A CapsuleNet-based framework for identifying DNA N6-methyladenine sites. Bioinformatics 2022, 38, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.; Chaudhary, T.; Eshaghzadeh Torbati, M.; Tudorascu, D.L.; Batmanghelich, K. ComBat Harmonization: Empirical Bayes versus Fully Bayes Approaches. NeuroImage Clin. 2023, 39, 103472. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.C.M.; Espadoto, M.; Hirata, R.; Telea, A.C. Constructing and Visualizing High-Quality Classifier Decision Boundary Maps. Information 2019, 10, 280. [Google Scholar] [CrossRef]
- Gerges, F.; Shih, F.; Azar, D. Automated diagnosis of acne and rosacea using convolution neural networks. In Proceedings of the 2021 4th International Conference on Artificial Intelligence and Pattern Recognition, Xiamen, China, 24–26 September 2021. [Google Scholar] [CrossRef]
- Hassan, H.F.; Koaik, L.; Khoury, A.E.; Atoui, A.; El Obeid, T.; Karam, L. Dietary exposure and risk assessment of mycotoxins in thyme and thyme-based products marketed in Lebanon. Toxins 2022, 14, 331. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.; Zeng, Q.; Wang, W.; Gadekallu, T.R.; Su, C. Blockchain-based two-stage federated learning with non-IID data in IoMT system. IEEE Trans. Comput. Soc. Syst. 2022, 10, 1701–1710. [Google Scholar] [CrossRef]
- Lee, J.-W. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 January 2021. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19-11-january-2021 (accessed on 6 November 2023).
- Rani, S.; Babbar, H.; Srivastava, G.; Gadekallu, T.R.; Dhiman, G. Security Framework for Internet of Things based Software Defined Networks using Blockchain. IEEE Internet Things J. 2022, 10, 6074–6081. [Google Scholar] [CrossRef]
- Zanca, F.; Hillis, S.L.; Claus, F.; Van Ongeval, C.; Celis, V.; Provoost, V.; Yoon, H.-J.; Bosmans, H. Correlation of free-response and receiver-operating-characteristic area-under-the-curve estimates: Results from independently conducted FROC/ROC studies in mammography. Med. Phys. 2012, 39, 5917–5929. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hyperparameters | Typical Values |
|---|---|
| E | 50 |
| TL | 4 |
| dʞ | 64 |
| Learning Rate (δ) | 0.001 |
| Batch Size | 64 |
| 0.5 | |
| 0.5 | |
| Epochs | 200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatesan, V.K.; Kuppusamy Murugesan, K.R.; Chandrasekaran, K.A.; Thyluru Ramakrishna, M.; Khan, S.B.; Almusharraf, A.; Albuali, A. Cancer Diagnosis through Contour Visualization of Gene Expression Leveraging Deep Learning Techniques. Diagnostics 2023, 13, 3452. https://doi.org/10.3390/diagnostics13223452
Venkatesan VK, Kuppusamy Murugesan KR, Chandrasekaran KA, Thyluru Ramakrishna M, Khan SB, Almusharraf A, Albuali A. Cancer Diagnosis through Contour Visualization of Gene Expression Leveraging Deep Learning Techniques. Diagnostics. 2023; 13(22):3452. https://doi.org/10.3390/diagnostics13223452
Chicago/Turabian StyleVenkatesan, Vinoth Kumar, Karthick Raghunath Kuppusamy Murugesan, Kaladevi Amarakundhi Chandrasekaran, Mahesh Thyluru Ramakrishna, Surbhi Bhatia Khan, Ahlam Almusharraf, and Abdullah Albuali. 2023. "Cancer Diagnosis through Contour Visualization of Gene Expression Leveraging Deep Learning Techniques" Diagnostics 13, no. 22: 3452. https://doi.org/10.3390/diagnostics13223452
APA StyleVenkatesan, V. K., Kuppusamy Murugesan, K. R., Chandrasekaran, K. A., Thyluru Ramakrishna, M., Khan, S. B., Almusharraf, A., & Albuali, A. (2023). Cancer Diagnosis through Contour Visualization of Gene Expression Leveraging Deep Learning Techniques. Diagnostics, 13(22), 3452. https://doi.org/10.3390/diagnostics13223452