
Molecular Testing in Ovarian Tumours: Challenges from the Pathologist’s Perspective


Abstract
1. Introduction
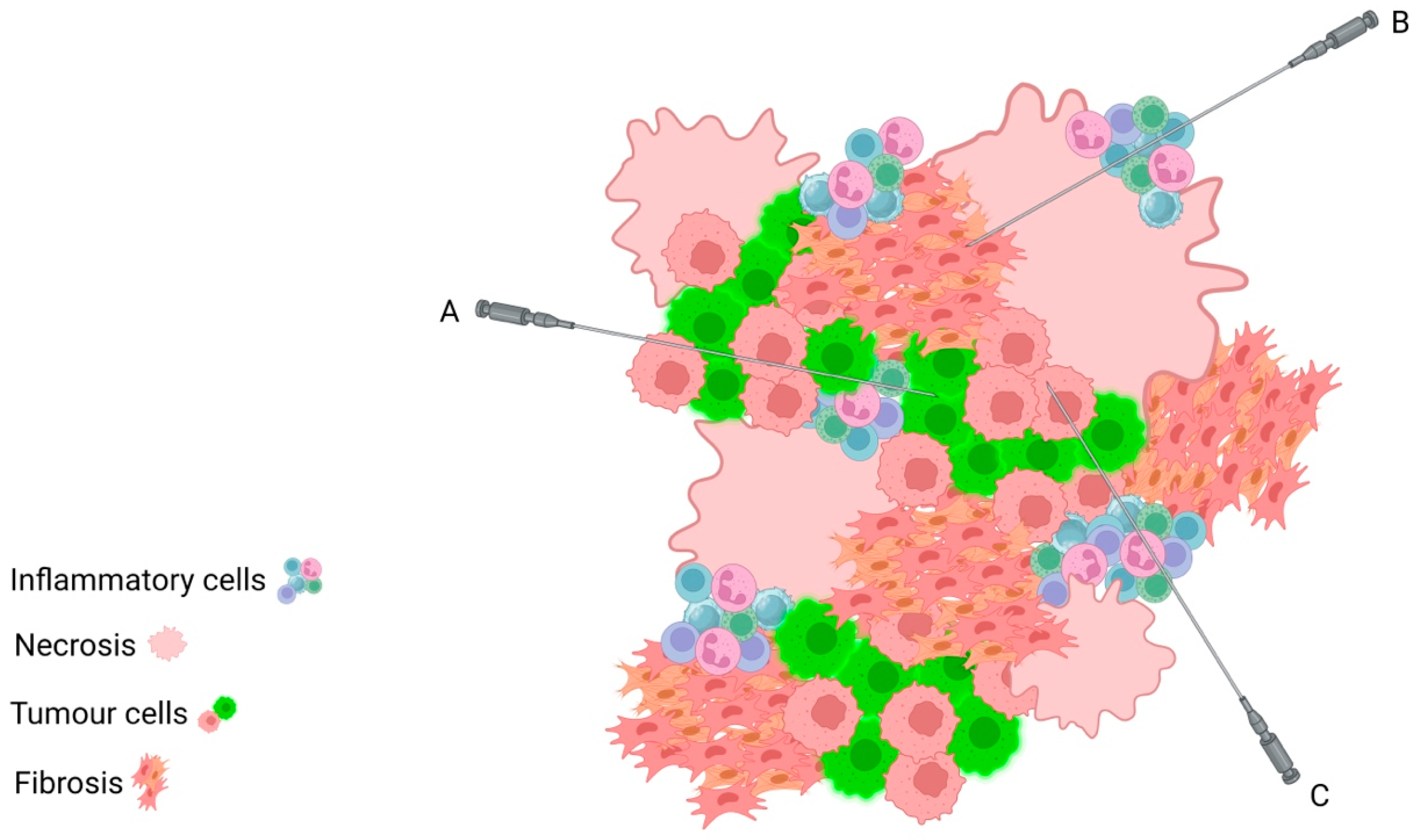
2. Considerations for Tissue Sampling and Interpretation
3. Pre-Analytical Considerations for the Pathologist
4. Post-Analytical Considerations for the Pathologist
5. Conclusions

{kind=link}
{kind=link}
| Tumour Type | Associated Molecular Aberrations |
|---|---|
| Epithelial Tumours | |
| High-grade serous carcinoma [7,12,38] | TP53 * HRD * Germline BRCA 1/2 * |
| Low-grade serous carcinoma [39] | KRAS BRAF NRAS |
| Mucinous carcinoma [26] | CDKN2A KRAS TP53 HER2 amplification |
| Endometrioid carcinoma [40,41,42] | MMR * CTNNB1 PIK3CA PTEN KRAS ARID1A POLE TP53 |
| Clear cell carcinoma [8,42,43,44,45] | MMR * ARID1A PIK3CA TERT KRAS TP53 |
| Brenner tumour [46] | Key molecular alterations not identified |
| Mesonephric-like adenocarcinoma [47,48] | PIK3CA KRAS NRAS |
| Carcinosarcoma [14] | TP53 * |
| Sex Cord Stromal Tumours | |
| Adult granulosa cell tumour [17] | FOXL2 (somatic) ** |
| Juvenile granulosa cell tumour [49,50,51] | AKT1 GNAS IDH 1/2 |
| Sertoli cell tumour [18] | DICER1 (somatic) ** |
| Sertoli–Leydig cell tumour [19,20,52] | DICER1 (somatic) ** FOXL2 |
| Sex cord tumour with annular tubules (SCTATs) [22] | STK11 ** |
| Germ Cell Tumours | |
| Yolk sac tumour [27] | Chromosome 12 abnormalities |
| Dysgerminoma [26] | Chromosome 12 abnormalities KIT |
| Embryonal carcinoma [28] | Chromosome 12 abnormalities |
| Mesenchymal Tumours | |
| Leiomyoma [24] | MED12 ** |
| Miscellaneous Tumours | |
| Small cell carcinoma of ovary, hypercalcaemic type [23] | SMARCA4 (somatic or germline) ** |
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.-H.; Bast, R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef]
- Guan, L.; Lu, Y. New developments in molecular targeted therapy of ovarian cancer. Discov. Med. 2018, 26, 219–229. [Google Scholar] [PubMed]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Fuh, K.; Mullen, M.; Blachut, B.; Stover, E.; Konstantinopoulos, P.; Liu, J.; Matulonis, U.; Khabele, D.; Mosammaparast, N.; Vindigni, A. Homologous recombination deficiency real-time clinical assays, ready or not? Gynecol. Oncol. 2020, 159, 877–886. [Google Scholar] [CrossRef]
- Frey, M.K.; Pothuri, B. Homologous recombination deficiency (HRD) testing in ovarian cancer clinical practice: A review of the literature. Gynecol. Oncol. Res. Pract. 2017, 4, 4. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Fraune, C.; Rosebrock, J.; Simon, R.; Hube-Magg, C.; Makrypidi-Fraune, G.; Kluth, M.; Büscheck, F.; Höflmayer, D.; Schmalfeldt, B.; Müller, V.; et al. High homogeneity of MMR deficiency in ovarian cancer. Gynecol. Oncol. 2020, 156, 669–675. [Google Scholar] [CrossRef]
- Dedeurwaerdere, F.; Claes, K.B.; Van Dorpe, J.; Rottiers, I.; Van der Meulen, J.; Breyne, J.; Swaerts, K.; Martens, G. Comparison of microsatellite instability detection by immunohistochemistry and molecular techniques in colorectal and endometrial cancer. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Parra-Herran, C.; Lerner-Ellis, J.; Xu, B.; Khalouei, S.; Bassiouny, D.; Cesari, M.; Ismiil, N.; Nofech-Mozes, S. Molecular-based classification algorithm for endometrial carcinoma categorizes ovarian endometrioid carcinoma into prognostically significant groups. Mod. Pathol. 2017, 30, 1748–1759. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Woo, H.Y.; Do, S.I.; Kim, H.S. Targeted sequencing of tubo-ovarian and peritoneal high-grade serous carcinoma with wild-type p53 immunostaining pattern. In Vivo 2019, 33, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Ogata, S.; Tamura, G.; Katayama, Y.; Fukase, M.; Yajima, M.; Motoyama, T. Carcinosarcomas (Malignant Mullerian Mixed Tumors) of the Uterus and Ovary: A Genetic Study With Special Reference to Histogenesis. Int. J. Gynecol. Pathol. 2003, 22, 368–373. [Google Scholar] [CrossRef]
- De Leo, A.; Santini, D.; Ceccarelli, C.; Santandrea, G.; Palicelli, A.; Acquaviva, G.; Chiarucci, F.; Rosini, F.; Ravegnini, G.; Pession, A.; et al. What Is New on Ovarian Carcinoma: Integrated Morphologic and Molecular Analysis Following the New 2020 World Health Organization Classification of Female Genital Tumors. Diagnostics 2021, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Cheasley, D.; Wakefield, M.J.; Ryland, G.L.; Allan, P.E.; Alsop, K.; Amarasinghe, K.C.; Ananda, S.; Anglesio, M.S.; Au-Yeung, G.; Böhm, M.; et al. The molecular origin and taxonomy of mucinous ovarian carcinoma. Nat. Commun. 2019, 10, 3935. [Google Scholar] [CrossRef]
- Shah, S.P.; Köbel, M.; Senz, J.; Morin, R.D.; Clarke, B.A.; Wiegand, K.C.; Leung, G.; Zayed, A.; Mehl, E.; Kalloger, S.E.; et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N. Engl. J. Med. 2009, 360, 2719–2729. [Google Scholar] [CrossRef]
- Conlon, N.; Schultheis, A.M.; Piscuoglio, S.; Silva, A.; Guerra, E.; Tornos, C.; E Reuter, V.; A Soslow, R.; Young, R.H.; Oliva, E.; et al. A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod. Pathol. 2015, 28, 1603–1612. [Google Scholar] [CrossRef]
- de Kock, L.; Terzic, T.; McCluggage, W.G.; Stewart, C.J.; Shaw, P.; Foulkes, W.D.; Clarke, B.A. DICER1 Mutations Are Consistently Present in Moderately and Poorly Differentiated Sertoli-Leydig Cell Tumors. Am. J. Surg. Pathol. 2017, 41, 1178–1187. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Yang, W.; Mo, F.; Senz, J.; Yap, D.; Anglesio, M.S.; Gilks, B.; Morin, G.B.; Huntsman, D.G. The Oncogenic Roles of DICER1 RNase IIIb Domain Mutations in Ovarian Sertoli-Leydig Cell Tumors. Neoplasia 2015, 17, 650–660. [Google Scholar] [CrossRef]
- Young, R.H.; Welch, W.R.; Dickersin, G.R.; Scully, R.E. Ovarian sex cord tumor with annular tubules: Review of 74 cases including 27 with Peutz-Jeghers syndrome and four with adenoma malignum of the cervix. Cancer 1982, 50, 1384–1402. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Mei, L.; Huang, Y.; Yang, P.; Li, H.; Peng, Y.; Chen, C.; Wei, X.; Pan, Q.; Liang, D.; et al. Three novel mutations of STK11 gene in Chinese patients with Peutz–Jeghers syndrome. BMC Med. Genet. 2016, 17, 77. [Google Scholar] [CrossRef]
- Jelinic, P.; Mueller, J.J.; Olvera, N.; Dao, F.; Scott, S.N.; Shah, R.; Gao, J.; Schultz, N.; Gonen, M.; A Soslow, R.; et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Maeda, D.; Kudo-Asabe, Y.; Tamura, D.; Nanjo, H.; Hayashi, A.; Ikemura, M.; Fukayama, M.; Goto, A. MED12 is frequently mutated in ovarian and other adnexal leiomyomas. Hum. Pathol. 2018, 81, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kaldawy, A.; Segev, Y.; Lavie, O.; Auslender, R.; Sopik, V.; Narod, S.A. Low-grade serous ovarian cancer: A review. Gynecol. Oncol. 2016, 143, 433–438. [Google Scholar] [CrossRef]
- Cheng, L.; Roth, L.M.; Zhang, S.; Wang, M.; Morton, M.J.; Zheng, W.; Karim, F.W.A.; Montironi, R.; Lopez-Beltran, A. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer 2010, 117, 2096–2103. [Google Scholar] [CrossRef]
- Riopel, M.A.; Spellerberg, A.; Griffin, C.A.; Perlman, E.J. Genetic analysis of ovarian germ cell tumors by comparative genomic hybridization. Cancer Res. 1998, 58, 3105–3110. [Google Scholar]
- Cheng, L.; Zhang, S.; Talerman, A.; Roth, L.M. Morphologic, immunohistochemical, and fluorescence in situ hybridization study of ovarian embryonal carcinoma with comparison to solid variant of yolk sac tumor and immature teratoma. Hum. Pathol. 2010, 41, 716–723. [Google Scholar] [CrossRef]
- Cheung, A.; Shah, S.; Parker, J.; Soor, P.; Limbu, A.; Sheriff, M.; Boussios, S. Non-Epithelial Ovarian Cancers: How Much Do We Really Know? Int. J. Environ. Res. Public Health 2022, 19, 1106. [Google Scholar] [CrossRef]
- Kossaï, M.; Leary, A.; Scoazec, J.Y.; Genestie, C. Ovarian cancer: A heterogeneous disease. Pathobiology 2018, 85, 41–49. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Lawson, B.C.; Euscher, E.D.; Bassett, R.L.; Liu, J.; Ramalingam, P.; Zhong, Y.; Fleming, N.D.; Malpica, A. A Three-Tier Chemotherapy Response Score For Ovarian/Fallopian Tube/Peritoneal High Grade Serous Carcinoma, Is It Clinically Relevant? Am. J. Surg. Pathol. 2020, 44, 206. [Google Scholar] [CrossRef] [PubMed]
- Sadeghipour, A.; Babaheidarian, P. Making formalin-fixed, paraffin embedded blocks. Biobanking 2018, 1897, 253–268. [Google Scholar]
- McDonough, S.J.; Bhagwate, A.; Sun, Z.; Wang, C.; Zschunke, M.; Gorman, J.A.; Kopp, K.J.; Cunningham, J.M. Use of FFPE-derived DNA in next generation sequencing: DNA extraction methods. PLoS ONE 2019, 14, e0211400. [Google Scholar] [CrossRef]
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef]
- Wallace, A.J. New challenges for BRCA testing: A view from the diagnostic laboratory. Eur. J. Hum. Genet. 2016, 24, S10–S18. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Tamayo, P.; Yang, J.-Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2012, 123, 517–525. [Google Scholar] [CrossRef]
- Hunter, S.M.; Anglesio, M.S.; Ryland, G.L.; Sharma, R.; Chiew, Y.-E.; Rowley, S.M.; Doyle, M.A.; Li, J.; Gilks, C.B.; Moss, P.; et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget 2015, 6, 37663–37677. [Google Scholar] [CrossRef]
- Wu, R.; Hendrix-Lucas, N.; Kuick, R.; Zhai, Y.; Schwartz, D.R.; Akyol, A.; Hanash, S.; Misek, D.E.; Katabuchi, H.; Williams, B.; et al. Mouse Model of Human Ovarian Endometrioid Adenocarcinoma Based on Somatic Defects in the Wnt/β-Catenin and PI3K/Pten Signaling Pathways. Cancer Cell 2007, 11, 321–333. [Google Scholar] [CrossRef] [PubMed]
- McConechy, M.K.; Ding, J.; Senz, J.; Yang, W.; Melnyk, N.; A Tone, A.; Prentice, L.M.; Wiegand, K.C.; McAlpine, J.N.; Shah, S.P.; et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod. Pathol. 2013, 27, 128–134. [Google Scholar] [CrossRef]
- Campbell, I.G.; Russell, S.E.; Choong, D.Y.H.; Montgomery, K.G.; Ciavarella, M.L.; Hooi, C.S.F.; Cristiano, B.E.; Pearson, R.B.; Phillips, W.A. Mutation of the PIK3CA Gene in Ovarian and Breast Cancer. Cancer Res. 2004, 64, 7678–7681. [Google Scholar] [CrossRef]
- Wu, R.-C.; Ayhan, A.; Maeda, D.; Kim, K.-R.; A Clarke, B.; Shaw, P.; Chui, M.H.; Rosen, B.; Shih, I.-M.; Wang, T.-L. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J. Pathol. 2014, 232, 473–481. [Google Scholar] [CrossRef]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 2017, 49, 856–865. [Google Scholar] [CrossRef]
- Parra-Herran, C.; Bassiouny, D.; Lerner-Ellis, J.; Olkhov-Mitsel, E.; Ismiil, N.; Hogen, L.; Vicus, D.; Nofech-Mozes, S. p53, mismatch repair protein, and POLE abnormalities in ovarian clear cell carcinoma: An outcome-based clinicopathologic analysis. Am. J. Surg. Pathol. 2019, 43, 1591–1599. [Google Scholar] [CrossRef]
- Folkins, A.; Palacios, J.; Cheng, X.W. WHO Classification of Tumours: Female Genital Tumours, 5th ed.; WHO Classification of Tumours Editorial Board: Geneva, Switzerland, 2020; Volume 4, ISBN 978-92-832-4504-9. [Google Scholar]
- Mirkovic, J.; McFarland, M.; Garcia, E.; Sholl, L.M.; Lindeman, N.; MacConaill, L.; Dong, F.; Hirsch, M.; Nucci, M.R.; Quick, C.M.; et al. Targeted Genomic Profiling Reveals Recurrent KRAS Mutations in Mesonephric-like Adenocarcinomas of the Female Genital Tract. Am. J. Surg. Pathol. 2018, 42, 227–233. [Google Scholar] [CrossRef]
- McCluggage, W.G.; Vosmikova, H.; Laco, J. Ovarian combined low-grade serous and mesonephric-like adenocarcinoma: Further evidence for a Mullerian origin of mesonephric-like adenocarcinoma. Int. J. Gynecol. Pathol. 2020, 39, 84–92. [Google Scholar] [CrossRef]
- Plevová, P.; Geržová, H. Genetic Causes of Rare Pediatric Ovarian Tumors. Klin. Onkol. 2019, 32 (Suppl. S2), 79–91. [Google Scholar] [CrossRef] [PubMed]
- Kalfa, N.; Ecochard, A.; Patte, C.; Duvillard, P.; Audran, F.; Pienkowski, C.; Thibaud, E.; Brauner, R.; Lecointre, C.; Plantaz, D.; et al. Activating mutations of the stimulatory g protein in juvenile ovarian granulosa cell tumors: A new prognostic factor? J. Clin. Endocrinol. Metab. 2006, 91, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Bessière, L.; Todeschini, A.-L.; Auguste, A.; Sarnacki, S.; Flatters, D.; Legois, B.; Sultan, C.; Kalfa, N.; Galmiche, L.; Veitia, R.A. A Hot-spot of In-frame Duplications Activates the Oncoprotein AKT1 in Juvenile Granulosa Cell Tumors. Ebiomedicine 2015, 2, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, A.N.; Wang, Y.; Keul, J.; Tessier-Cloutier, B.; Magrill, J.; Kommoss, S.; Senz, J.; Yang, W.; Proctor, L.; Schmidt, D.; et al. DICER1 and FOXL2 Mutation Status Correlates With Clinicopathologic Features in Ovarian Sertoli-Leydig Cell Tumors. Am. J. Surg. Pathol. 2019, 43, 628–638. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinneen, K.; Arora, R. Molecular Testing in Ovarian Tumours: Challenges from the Pathologist’s Perspective. Diagnostics 2023, 13, 2072. https://doi.org/10.3390/diagnostics13122072
Dinneen K, Arora R. Molecular Testing in Ovarian Tumours: Challenges from the Pathologist’s Perspective. Diagnostics. 2023; 13(12):2072. https://doi.org/10.3390/diagnostics13122072
Chicago/Turabian StyleDinneen, Kate, and Rupali Arora. 2023. "Molecular Testing in Ovarian Tumours: Challenges from the Pathologist’s Perspective" Diagnostics 13, no. 12: 2072. https://doi.org/10.3390/diagnostics13122072
APA StyleDinneen, K., & Arora, R. (2023). Molecular Testing in Ovarian Tumours: Challenges from the Pathologist’s Perspective. Diagnostics, 13(12), 2072. https://doi.org/10.3390/diagnostics13122072