
Complete Genomic Characterisation and Mutation Patterns of Iraqi SARS-CoV-2 Isolates


Abstract
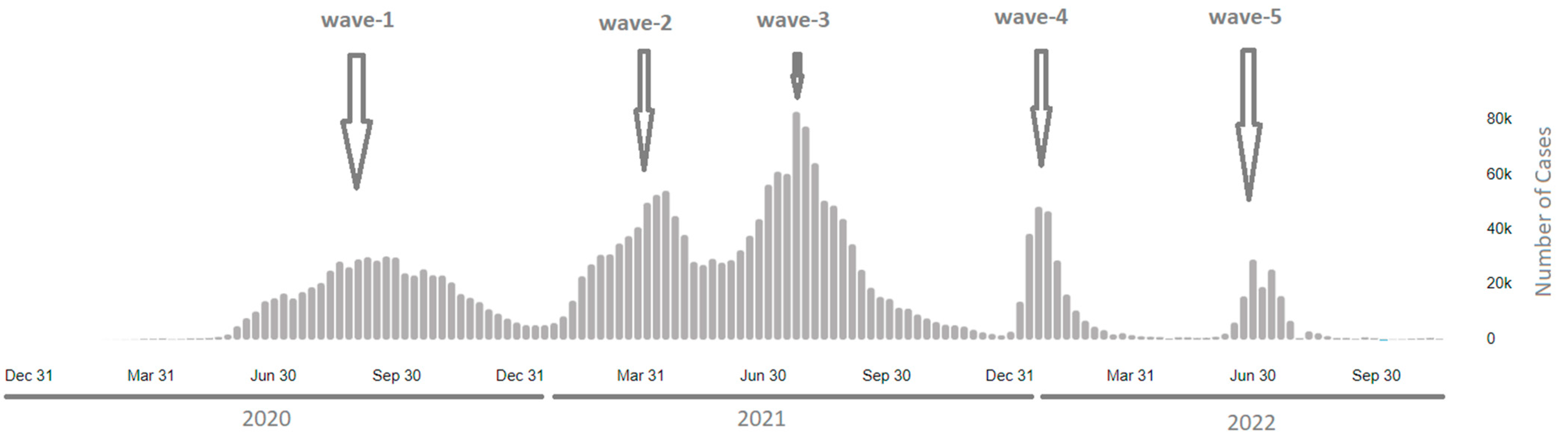
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Viral RNA Extraction and Real-Time PCR
2.3. SARS-CoV-2 Next-Generation Sequencing (NGS)
2.4. Bioinformatics Analysis
2.5. Lineage and Phylogenetic Analysis
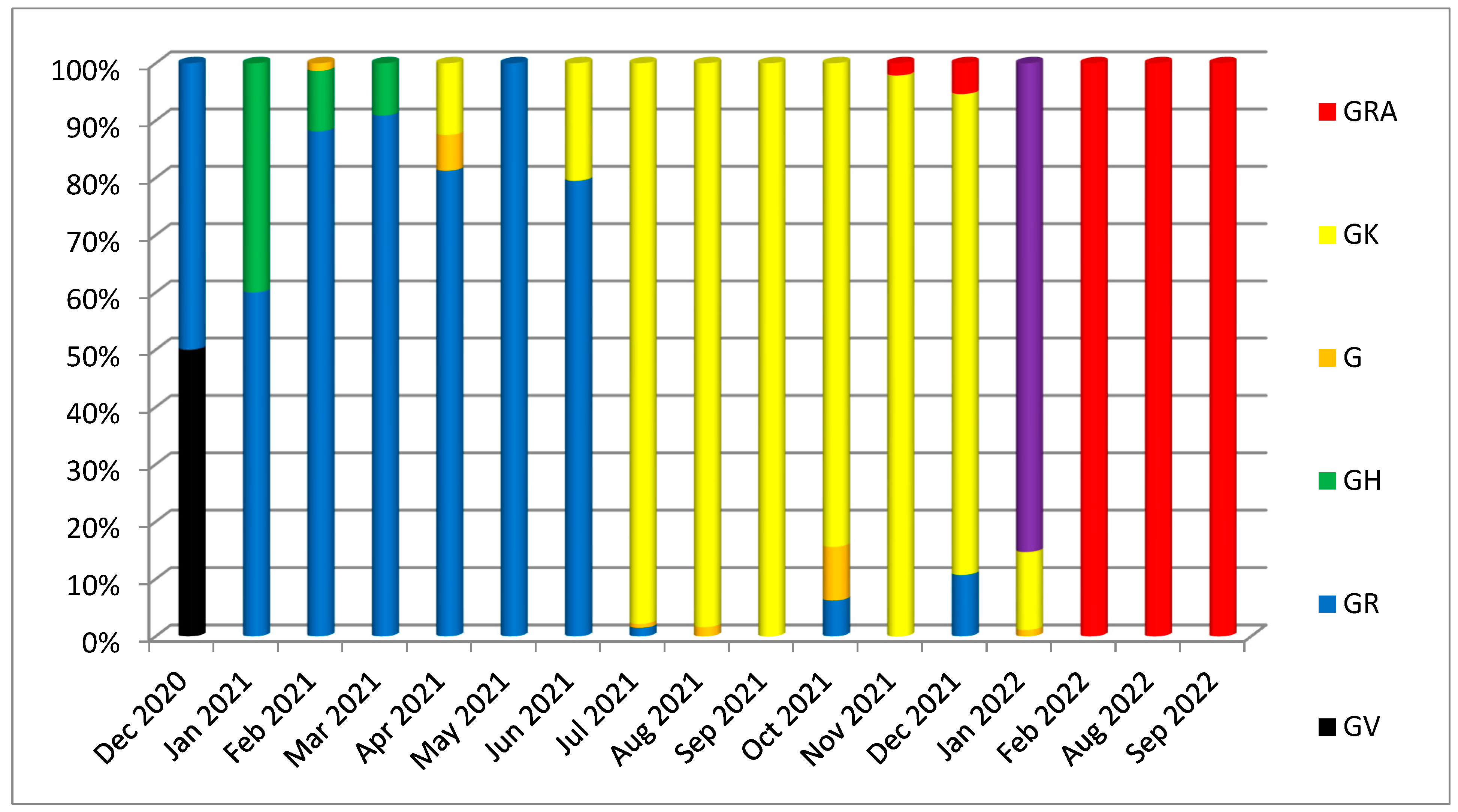
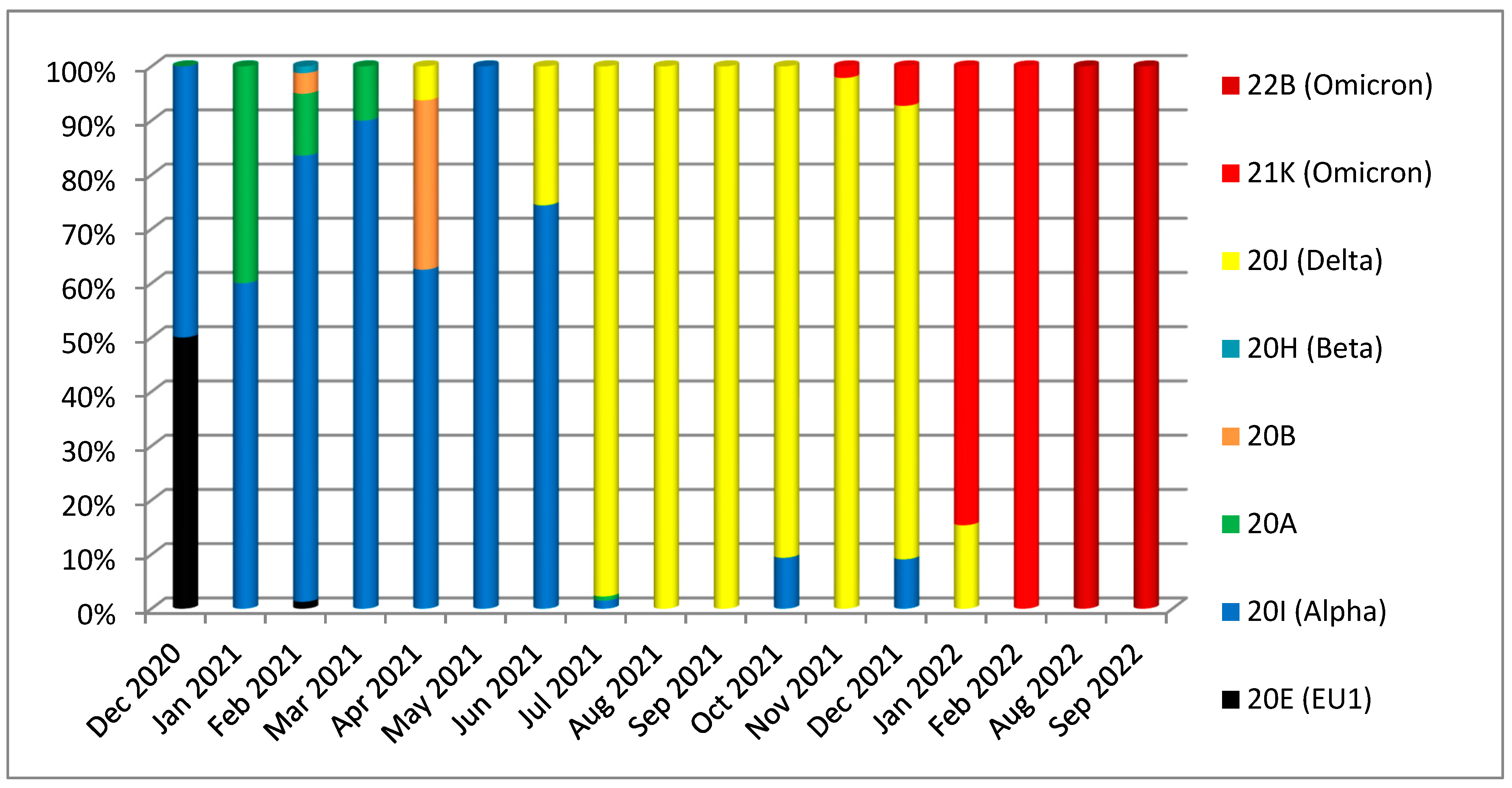
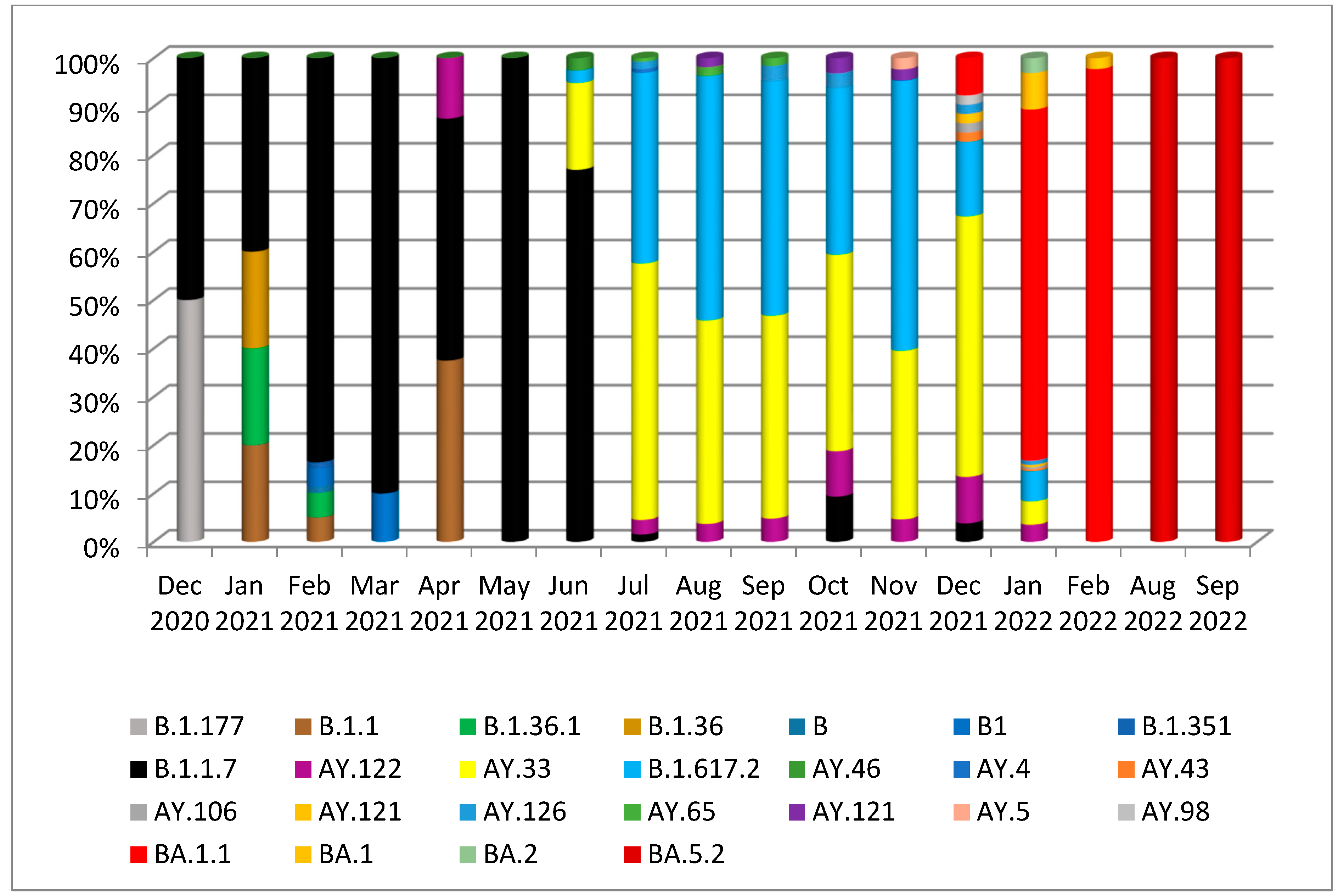
3. Results
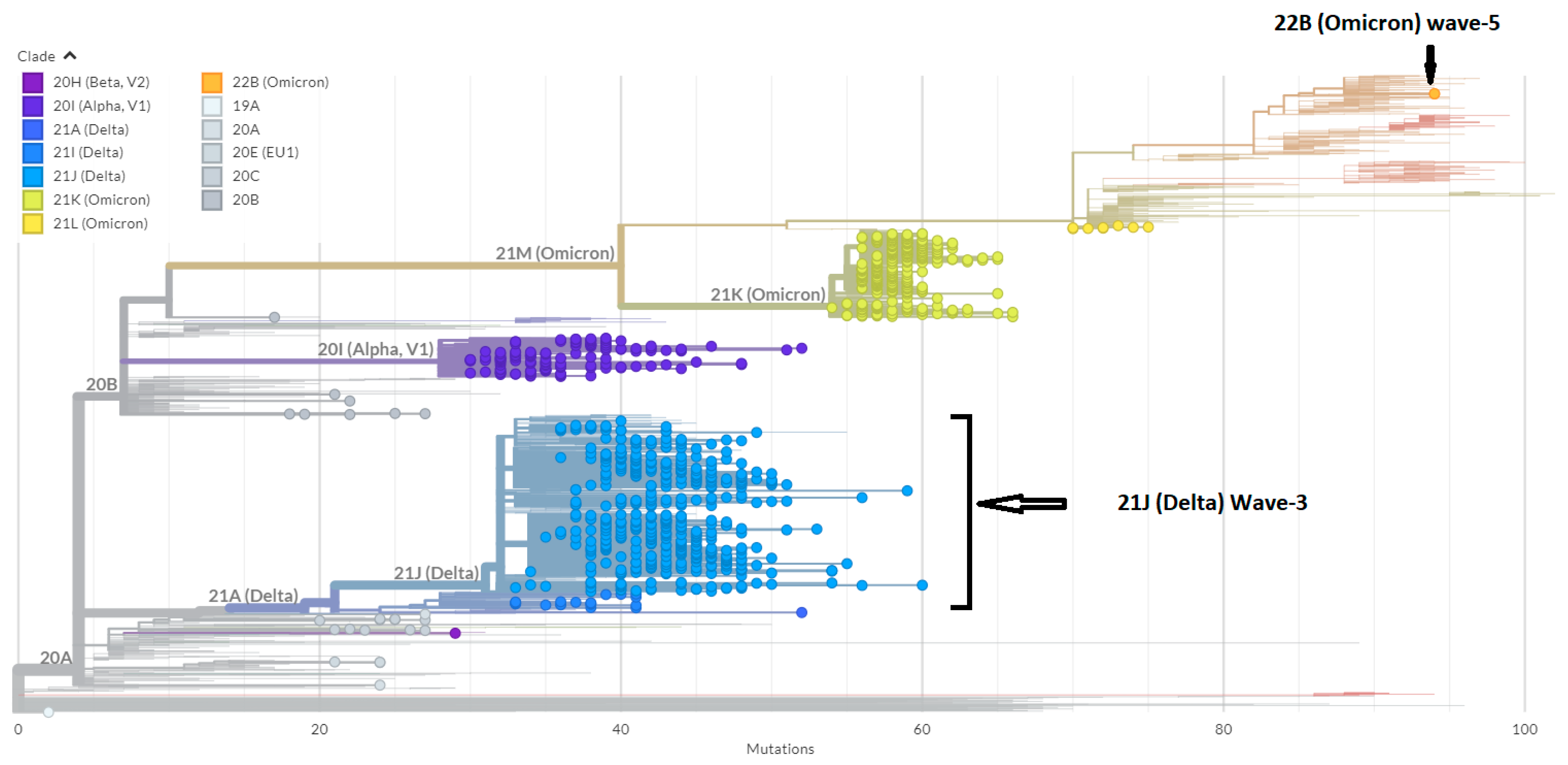
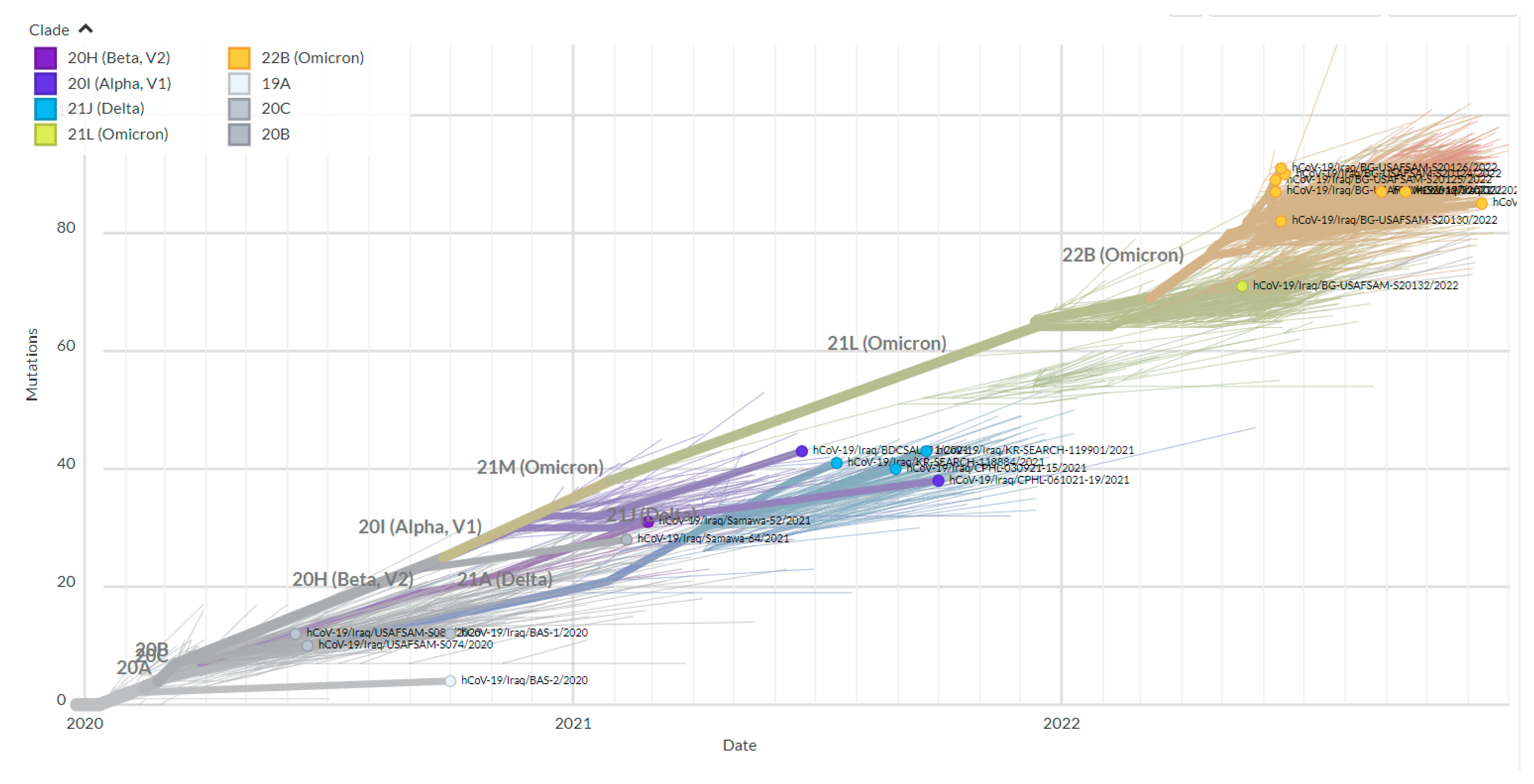
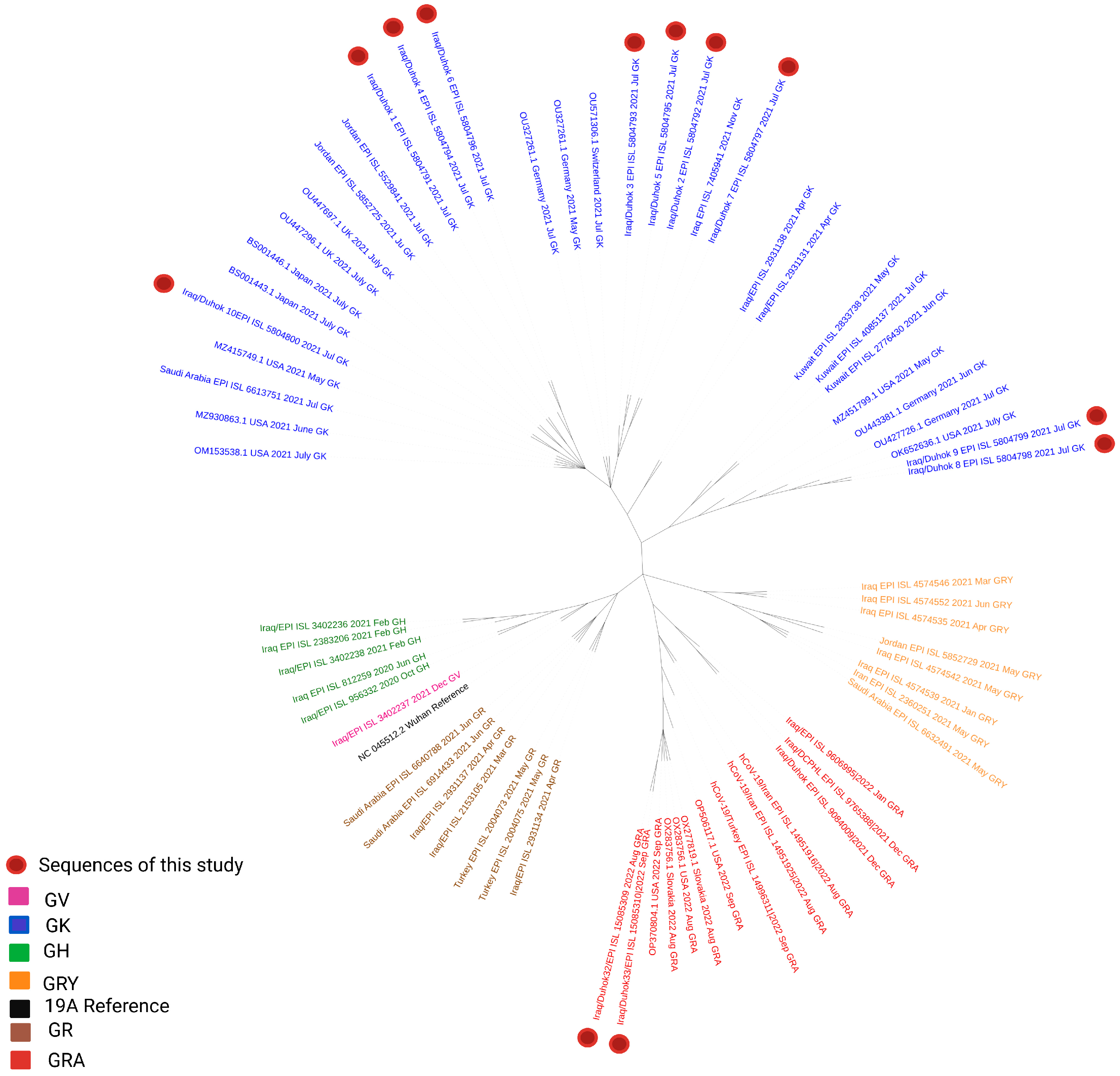
3.1. Phylogenetic Analysis of the Third Wave Isolates and Other Iraqi Sequences
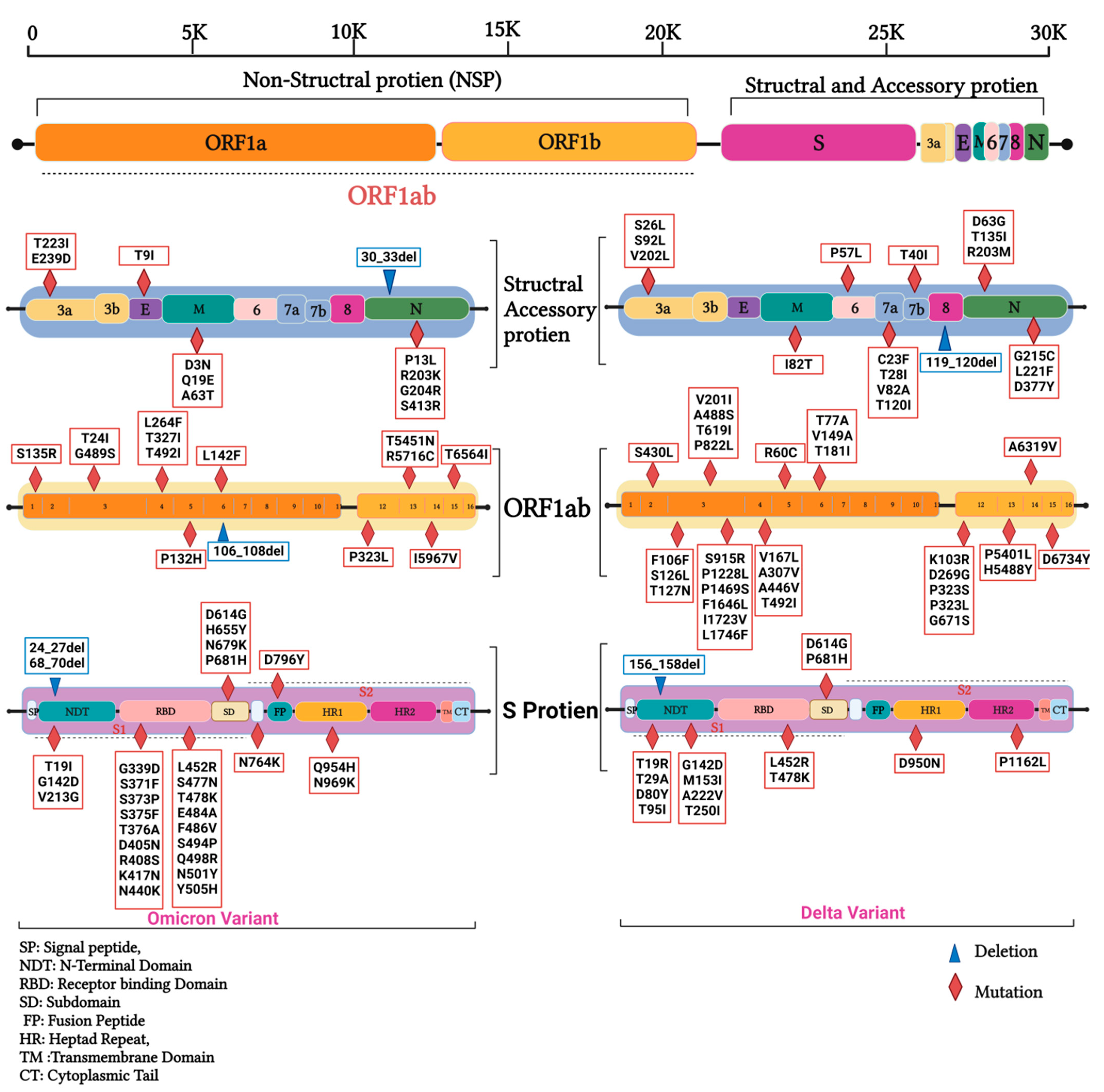
3.2. Mutations in SARS-CoV2 Genomes of the Duhok Isolates during the Third Wave of the Pandemic in Iraq
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. Atenei Parm. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. WHO Coronavirus (COVID-19) Dashboard With Vaccination Data. WHO, 2021; pp. 1–5. Available online: https://covid19.who.int/ (accessed on 21 November 2022).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Challenges 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Miljanovic, D.; Milicevic, O.; Loncar, A.; Abazovic, D.; Despot, D.; Banko, A. The First Molecular Characterization of Serbian SARS-CoV-2 Isolates From a Unique Early Second Wave in Europe. Front. Microbiol. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Kim, J.-S.; Jang, J.-H.; Kim, J.-M.; Chung, Y.-S.; Yoo, C.-K.; Han, M.-G. Genome-Wide Identification and Characterization of Point Mutations in the SARS-CoV-2 Genome. Osong Public Health Res. Perspect. 2020, 11, 101–111. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Ahmad, J.; Tayib, G.; Mohamed, T. SARS-CoV-2 mutation hotspots incidence in different geographic regions. Microb. Biosyst. 2020, 5, 1–8. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High Fidelity of Murine Hepatitis Virus Replication Is Decreased in nsp14 Exoribonuclease Mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef]
- Amicone, M.; Borges, V.; João Alves, M.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Paulo Gomes, J.; Gordo, I.; et al. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. BiorXiv 2021, 10, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Goeman, J.J.; de Parquet, M.C.; Thi Nga, P.; Snijder, E.J.; Morita, K.; Gorbalenya, A.E. The Footprint of Genome Architecture in the Largest Genome Expansion in RNA Viruses. PLoS Pathog. 2013, 9, e1003500. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, O.P.; Priyanka; Ali, R.K.; Maulud, S.Q.; Dhawan, M.; Mohammed, T.A. Will the next spillover pandemic be deadlier than the COVID-19?: A wake-up call. Int. J. Surg. 2022, 97, 106208. [Google Scholar] [CrossRef] [PubMed]
- Molina-Mora, J.A.; Cordero-Laurent, E.; Godínez, A.; Calderón-Osorno, M.; Brenes, H.; Soto-Garita, C.; Pérez-Corrales, C.; Drexler, J.F.; Moreira-Soto, A.; Corrales-Aguilar, E.; et al. SARS-CoV-2 genomic surveillance in Costa Rica: Evidence of a divergent population and an increased detection of a spike T1117I mutation. Infect. Genet. Evol. 2021, 92, 104872. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, O.P.; Priyanka; Ahmed, J.Q.; Mohammed, T.A.; Singh, I.; Rodriguez-Morales, A.J. Heterologous prime-boost vaccination against COVID-19: Is it safe and reliable? Hum. Vaccin. Immunother. 2021, 17, 5135–5138. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Pathirana, P.N.; Nguyen, T.; Nguyen, Q.V.H.; Bhatti, A.; Nguyen, D.C.; Nguyen, D.T.; Nguyen, N.D.; Creighton, D.; Abdelrazek, M. Genomic mutations and changes in protein secondary structure and solvent accessibility of SARS-CoV-2 (COVID-19 virus). Sci. Rep. 2021, 11, 3487. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Oxford Univ. Press 2013, arXiv:1303.3997. Available online: https://arxiv.org/abs/1303.3997 (accessed on 21 November 2022).
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Yuelong, S.; John, M. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro surveill. 2017, 22, 30494. [Google Scholar] [CrossRef]
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; Van Kerkhove, M.D.; Jombart, T.; et al. Increased transmissibility and global spread of SARSCoV- 2 variants of concern as at June 2021. Eurosurveillance 2021, 26, 1–6. [Google Scholar] [CrossRef]
- Grant, R.; Charmet, T.; Schaeffer, L.; Galmiche, S.; Madec, Y.; Von Platen, C.; Chény, O.; Omar, F.; David, C.; Rogoff, A.; et al. Impact of SARS-CoV-2 Delta variant on incubation, transmission settings and vaccine effectiveness: Results from a nationwide case-control study in France. Lancet Reg. Health Eur. 2021, 13, 100278. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Ahmed, J.Q.; Maulud, S.Q.; Al-Qadi, R.; Mohamed, T.A.; Tayib, G.A.; Hassan, A.M.; Taha, L.S.; Qasim, K.M.; Tawfeeq, M.A. Sequencing and mutations analysis of the first recorded SARS-CoV-2 Omicron variant during the fourth wave of pandemic in Iraq. Brazilian J. Infect. Dis. 2022, 26, 102677. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Joh, E.; Buchan, S.A.; Daneman, N.; Mishra, S.; Patel, S.; Day, T. Inflection in prevalence of SARS-CoV-2 infections missing the N501Y mutation as a marker of rapid Delta (B.1.617.2) lineage expansion in Ontario, Canada. MedRxiv 2021. Available online: https://doi.org/10.1101/2021.06.22.21259349 (accessed on 21 November 2022).
- Domingo, E.; García-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Irwin, K.K.; Renzette, N.; Kowalik, T.F.; Jensen, J.D. Antiviral drug resistance as an adaptive process. Virus Evol. 2016, 2, vew014. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Subissi, L.; Silveira De Morais, A.T.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. 2018, 115, E162–E171. [Google Scholar] [CrossRef]
- Hatirnaz Ng, O.; Akyoney, S.; Sahin, I.; Soykam, H.O.; Bayram Akcapinar, G.; Ozdemir, O.; Kancagi, D.D.; Sir Karakus, G.; Yurtsever, B.; Kocagoz, A.S.; et al. Mutational landscape of SARS-CoV-2 genome in Turkey and impact of mutations on spike protein structure. PLoS ONE 2021, 16, e0260438. [Google Scholar] [CrossRef]
- Vidanović, D.; Tešović, B.; Volkening, J.D.; Afonso, C.L.; Quick, J.; Šekler, M.; Knežević, A.; Janković, M.; Jovanović, T.; Petrović, T.; et al. First whole-genome analysis of the novel coronavirus (SARS-CoV-2) obtained from COVID-19 patients from five districts in Western Serbia. Epidemiol. Infect. 2021, 149, e246. [Google Scholar] [CrossRef]
- Sakai, Y.; Kawachi, K.; Terada, Y.; Omori, H.; Matsuura, Y.; Kamitani, W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology 2017, 510, 165–174. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- McBride, R.; van Zyl, M.; Fielding, B. The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid Protein Recruitment to Replication-Transcription Complexes Plays a Crucial Role in Coronaviral Life Cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sasi, S.; Pillai, S.G.; Nag, A.; Shukla, D.; Singhal, R.; Phalke, S.; Velu, G.S.K. SARS-CoV-2 Mutations and Their Impact on Diagnostics, Therapeutics and Vaccines. Front. Med. 2022, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Bozdayi, G.; Yigit, S.; Muftah, H.; Dizbay, M.; Tunccan, O.G.; Fidan, I.; Caglar, K. Genomic characterization of SARS-CoV-2 isolates from patients in Turkey reveals the presence of novel mutations in spike and nsp12 proteins. J. Med. Virol. 2021, 93, 6016–6026. [Google Scholar] [CrossRef]
- Ko, K.; Nagashima, S.; Bunthen, E.; Ouoba, S.; Akita, T.; Sugiyama, A.; Ohisa, M.; Sakaguchi, T.; Tahara, H.; Ohge, H.; et al. Molecular characterization and the mutation pattern of SARS-CoV-2 during first and second wave outbreaks in Hiroshima, Japan. PLoS ONE 2021, 16, e0246383. [Google Scholar] [CrossRef]
- Rahman, M.M.; Kader, S.B.; Rizvi, S.M.S. Molecular characterization of SARS-CoV-2 from Bangladesh: Implications in genetic diversity, possible origin of the virus, and functional significance of the mutations. Heliyon 2021, 7, e07866. [Google Scholar] [CrossRef]
- Shiehzadegan, S.; Alaghemand, N.; Fox, M.; Venketaraman, V. Analysis of the Delta Variant B.1.617.2 COVID-19. Clin. Pract. 2021, 11, 778–784. [Google Scholar] [CrossRef]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe Acute Respiratory Syndrome Coronavirus ORF6 Antagonizes STAT1 Function by Sequestering Nuclear Import Factors on the Rough Endoplasmic Reticulum/Golgi Membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [CrossRef]
- Zekri, A.-R.N.; Easa Amer, K.; Hafez, M.M.; Hassan, Z.K.; Ahmed, O.S.; Soliman, H.K.; Bahnasy, A.A.; Abdel Hamid, W.; Gad, A.; Ali, M.; et al. Genomic characterization of SARS-CoV-2 in Egypt. J. Adv. Res. 2021, 30, 123–132. [Google Scholar] [CrossRef]
- Parham, K.A.; Kim, G.N.; Saeedian, N.; Ninkov, M.; Richer, C.G.; Li, Y.; Wu, K.; Rashu, R.; Barr, S.D.; Arts, E.J.; et al. Monovalent and trivalent VSV-based COVID-19 vaccines elicit potent neutralizing antibodies and immunodominant CD8+ T cells against diverse SARS-CoV-2 variants. BioRxiv 2022. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Nonsynonymous Mutations | Synonymous Mutations | Frameshift Deletion /Non-Frame Deletion | Upstream/Downstream Mutations | Total | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Delta | Omicron | Delta | Omicron | Delta | Omicron | Delta | Omicron | Delta | Omicron | |
| 5’UTR | – | – | – | – | – | – | 3 | 2 | 3 | 2 |
| ORF1a | 22 | 8 | 14 | 11 | – | 1 | – | – | 36 | 20 |
| ORF1b | 10 | 5 | 7 | 2 | – | – | – | – | 17 | 7 |
| S | 14 | 29 | 7 | 1 | 1 | 2 | – | – | 22 | 32 |
| ORF3a | 3 | 2 | 1 | 2 | – | – | – | – | 4 | 4 |
| E | – | 1 | – | – | – | – | – | – | 0 | 1 |
| M | 1 | 3 | 1 | 1 | – | – | – | – | 2 | 4 |
| ORF6 | 1 | – | – | – | – | – | – | – | 1 | 0 |
| ORF7a | 4 | – | – | 1 | – | – | – | – | 4 | 1 |
| ORF7b | 1 | – | – | 1 | – | – | – | – | 1 | 1 |
| ORF8 | – | – | – | – | 1 | – | – | – | 1 | 0 |
| N | 6 | 5 | 1 | 1 | – | 1 | – | – | 7 | 7 |
| ORF10 | 1 | – | – | – | – | – | – | – | 1 | 0 |
| 3’UTR | – | – | – | – | – | – | 3 | 1 | 3 | 1 |
| Total | 63 (61.7%) | 53(70%) | 31 (30.3%) | 20 (25%) | 2 | 4 | 6 | 3 | 102 | 80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, J.Q.; Maulud, S.Q. Complete Genomic Characterisation and Mutation Patterns of Iraqi SARS-CoV-2 Isolates. Diagnostics 2023, 13, 8. https://doi.org/10.3390/diagnostics13010008
Ahmed JQ, Maulud SQ. Complete Genomic Characterisation and Mutation Patterns of Iraqi SARS-CoV-2 Isolates. Diagnostics. 2023; 13(1):8. https://doi.org/10.3390/diagnostics13010008
Chicago/Turabian StyleAhmed, Jivan Qasim, and Sazan Qadir Maulud. 2023. "Complete Genomic Characterisation and Mutation Patterns of Iraqi SARS-CoV-2 Isolates" Diagnostics 13, no. 1: 8. https://doi.org/10.3390/diagnostics13010008
APA StyleAhmed, J. Q., & Maulud, S. Q. (2023). Complete Genomic Characterisation and Mutation Patterns of Iraqi SARS-CoV-2 Isolates. Diagnostics, 13(1), 8. https://doi.org/10.3390/diagnostics13010008