
A New Method for the Assessment of Myalgia in Interstitial Lung Disease: Association with Positivity for Myositis-Specific and Myositis-Associated Antibodies

 ,
,  , , ,
, , ,  ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Main Outcome Variable
2.3. Procedures
2.3.1. Laboratory Assessment
2.3.2. High-Resolution Computed Tomography
2.3.3. Complementary Exams
2.4. Diagnosis of Autoimmune Conditions
2.5. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Saketoo, L.A.; Ascherman, D.P.; Cottin, V.; Christopher-Stine, L.; Danoff, S.K.; Oddis, C.V. Interstitial Lung Disease in Idiopathic Inflammatory Myopathy. Curr. Rheumatol. 2020, 6, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Cavagna, L.; Trallero-Araguás, E.; Meloni, F.; Cavazzana, I.; Rojas-Serrano, J.; Feist, E.; Zanframundo, G.; Morandi, V.; Meyer, A.; da Silva, J.A.P.; et al. Influence of Antisynthetase Antibodies Specificities on Antisynthetase Syndrome Clinical Spectrum Time Course. J. Clin. Med. 2019, 8, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palterer, B.; Vitiello, G.; Carraresi, A.; Giudizi, M.G.; Cammelli, D.; Parronchi, P. Bench to bedside review of myositis autoantibodies. Clin. Mol. Allergy 2018, 16, 5. [Google Scholar] [CrossRef] [Green Version]
- Hengstman, G.J.D.; Brouwer, R.; Vree Egberts, W.T.M.; Seelig, H.P.; Jongen, P.J.H.; Van Venrooij, W.J.; Van Engelen, B.G.M. Clinical and serological characteristics of 125 Dutch myositis patients. J. Neurol. 2002, 249, 65–75. [Google Scholar] [CrossRef]
- Noda, K.; Yoshida, K.; Ukichi, T.; Furuya, K.; Hirai, K.; Kingetsu, I.; Kurosaka, D. Myalgia in Patients with Dermatomyositis and Polymyositis Is Attributable to Fasciitis Rather Than Myositis: A Retrospective Study of 32 Patients who Underwent Histopathological Examinations. J. Rheumatol. 2017, 44, 482–487. [Google Scholar] [CrossRef]
- Fischer, A.; Antoniou, K.M.; Brown, K.K.; Cadranel, J.; Corte, T.J.; Du Bois, R.M.; Lee, J.S.; Leslie, K.O.; Lynch, D.A.; Matteson, E.L.; et al. An official European Respiratory Society/American Thoracic Society research statement: Interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015, 46, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Connors, G.R.; Christopher-Stine, L.; Oddis, C.B.; Danoff, S.K. Interstitial Lung Disease associated with the idiopathic inflammatory myopathies: What progress has been made in the past 35 years? Chest 2010, 138, 1464–1474. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, I.E.; Tjärnlund, A.; Bottai, M.; Werth, V.P.; Pilkington, C.; De Visser, M.; Alfredsson, L.; Amato, A.A.; Barohn, R.J.; Liang, M.H.; et al. 2017 European Leaguea Against Rheumatism/American College of Rheumatology classification criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and their major subgroups. Ann. Rheum Dis. 2017, 69, 2271–2282. [Google Scholar] [CrossRef]
- Hervier, B.; Uzunhan, Y. Inflammatory Myopathy-Related Interstitial Lung Disease: From pathophysiology to treatment. Front. Med. 2020, 6, 326. [Google Scholar] [CrossRef]
- Drent, M.; Cobben, N.A.; Henderson, R.F.; Wouters, E.F.; Van Dieijen-Visser, M. Usefulness of lactate dehydrogenase and its isoenzymes as indicators of lung damage or inflammation. Eur. Respir. J. 1996, 9, 1736–1742. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Collard, H.R.; Raghu, G.; Sweet, M.P.; Hays, S.R.; Campos, G.M.; Golden, J.A.; King, T.E., Jr. Does Chronic Microaspiration cause Idiopathic Pulmonary Fibrosis? Am. J. Med. 2010, 123, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Sambataro, G.; Sambataro, D.; Pignataro, F.; Torrisi, S.E.; Vancheri, A.; Pavone, M.; Palmucci, S.; Del Papa, N.; Vancheri, C. Interstitial Lung Disease in patients with Polymyalgia Rheumatica: A case series. Respir. Med. Case Rep. 2018, 26, 126–130. [Google Scholar] [CrossRef]
- Douglass-Molloy, H.; Limaye, V. Prevalence of polymyalgia reumatica in a cohort of patients with idiopathic inflammatory myopathy. Clin. Rheumatol. 2020, 39, 1217–1221. [Google Scholar] [CrossRef]
- Tiniakou, E.; Mammen, A.L. Idiopathic Inflammatory Myopathies and malignancy: A comprehensive review. Clin. Rev. Allergy Immunol. 2017, 52, 20–33. [Google Scholar] [CrossRef]
- Sambataro, D.; Sambataro, G.; Pignataro, F.; Zanframundo, G.; Codullo, V.; Fagone, E.; Martorana, E.; Ferro, F.; Orlandi, M.; Del Papa, N.; et al. Patients with Interstitial Lung Disease Secondary to Autoimmune Diseases: How to recognize them? Diagnostics 2020, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Chang, C.; Lian, Z.X. Polymyositis and dermatomyositis- challenges in diagnosis and management. J. Transl. Autoimmun. 2019, 2, 100018. [Google Scholar] [CrossRef]
- Satoh, M.; Tanaka, S.; Ceribelli, A.; Calise, S.J.; Chan, E.K.L. A comprehensive overview on Myositis-Specific Antibodies: New and old biomarkers in Idiopathic Inflammatory Myopathy. Clin. Rev. Allergy Immunol. 2017, 52, 1–19. [Google Scholar] [CrossRef] [Green Version]
- American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir Crit Care Med. 2002, 165, 277–304. [Google Scholar] [CrossRef] [Green Version]
- Cutolo, M.; Sulli, A.; Smith, V. How to perform and interpret capillaroscopy. Best Pract. Res. Clin. Rheumatol. 2013, 27, 237–248. [Google Scholar] [CrossRef]
- Sambataro, D.; Sambataro, G.; Libra, A.; Vignigni, G.; Pino, F.; Fagone, E.; Fruciano, M.; Gili, E.; Pignataro, F.; Del Papa, N.; et al. Nailfold Videocapillaroscopy is a useful tool to recognize definite forms of systemic sclerosis and Idiopathic Inflammatory Myositis in Interstitial Lung Disease patients. Diagnostics 2020, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, M.; Triantafyllias, K.; Manfredi, A.; González-Gay, M.A.; Palmou-Fontana, N.; Cassone, G.; Drott, U.; Delbrück, C.; Rojas-Serrano, J.; Bertolazzi, C.; et al. Nailfold Capillaroscopy characteristics of Antisynthetase Syndrome and possible clinical associations: Results of a Multicenter International Study. J. Rheumatol. 2019, 46, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guidelines. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2013, 65, 2737–2747. [Google Scholar] [CrossRef] [Green Version]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against rheumatism classification criteria for Primary Sjögren’s Syndrome: A consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017, 69, 9–16. [Google Scholar] [CrossRef]
- Bohan, A.; Peter, J.B. Polymyositis and dermatomyositis (first of two parts). N. Engl. J. Med. 1975, 292, 344–347. [Google Scholar] [CrossRef]
- Bohan, A.; Peter, J.B. Polymyositis and dermatomyositis (second of two parts). N. Engl. J. Med. 1975, 292, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Hernandez, O.D.; Shoenfeld, Y. Mixed connective tissue disease: An overview of clinical manifestations, diagnosis and treatment. Best Pract. Res. Clin. Rheumatol. 2012, 26, 61–72. [Google Scholar] [CrossRef]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 EULAR/ACR Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, S. ANCA-associated vasculitis: Diagnostic and therapeutic strategy. Allergol. Int. 2007, 56, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guidelines. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Furini, F.; Carnevale, A.; Casoni, G.L.; Guerrini, G.; Cavagna, L.; Govoni, M.; Scirè, C.A. The role of the Multidisciplinary evaluation of interstitial lung diseases: Systematic literature review of the current evidence and future perspectives. Front. Med. 2019, 6, 246. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.H.; Kim, Y.J.; Kim, J.J.; Choi, C.B.; Sung, Y.K.; Kim, T.H.; Jun, J.B.; Bae, S.C.; Yoo, D.H. Mortality factors in idiopathic inflammatory myopathy: Focusing on malignancy and interstitial lung disease. Mod. Rheumatol. 2013, 23, 503–508. [Google Scholar] [CrossRef]
- Wang, S.M.; Han, C.; Lee, S.J.; Patkar, A.A.; Masand, P.S.; Pae, C.U. Fibromyalgia diagnosis: A review of the past, present and future. Expert Rev. Neurother. 2015, 15, 667–679. [Google Scholar] [CrossRef]
- Ostrovrsnik, J.; Hocevar, A.; Rotar, Z.; Krosel, M.; Cucnik, S.; Jurcic, V.; Tomsic, M. The incidence of idiopathic inflammatory myopathies in the adult Slovenian population. Clin. Rheumatol. 2019, 38, 279–283. [Google Scholar] [CrossRef]
- Narvaez, J.; Sanchez-Fernandez, S.A.; Seoane-Mato, D.; Diaz-Gonzalez, F.; Bustabad, S. Prevalence of Sjogren’s syndrome in the general adult population in Spain: Estimating the proportion of undiagnosed cases. Sci Rep. 2020, 10, 10627. [Google Scholar] [CrossRef]
- Choi, B.U.; Oh, H.J.; Lee, Y.J.; Song, Y.W. Prevalence and clinical impact of fibromyalgia in patients with primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2016, 34, S9–S13. [Google Scholar]
- Gau, S.Y.; Leong, P.Y.; Lin, C.L.; Tsou, H.K.; Wei, J.C.C. Higher risk for Sjogren’s syndrome patients with fibromyalgia: A nationwide population-based cohort study. Front. Immunol. 2021, 12, 640618. [Google Scholar] [CrossRef]
- Vulsteke, J.B.; Bossuyt, X.; Dillaerts, D.; Poesen, K.; Claeys, K.; Lenaerts, J.; Westhovens, R.; Blockmans, D.; De Haes, P.; De Langhe, E. FRI0393 Prevalence of myositis-specific antibodies in idiopathic inflammatory myopathy compared to disease and healthy controls [abstract]. Ann. Rheum Dis. 2017, 6, 636–637. [Google Scholar] [CrossRef]
- Espinosa-Ortega, F.; Holmqvist, M.; Alexanderson, H.; Stofors, H.; Mimori, T.; Lundberg, I.E.; Ronnelid, J. Comparison of autoantibody specificities tested by a line blot assay and immunoprecipitation-based algorithm in patients with idiopathic inflammatory myopathies. Ann. Rheum Dis. 2019, 78, 858–860. [Google Scholar] [CrossRef]
- Sambataro, G.; Ferro, F.; Orlandi, M.; Sambataro, D.; Torrisi, S.E.; Quartuccio, L.; Vancheri, C.; Baldini, C.; Matucci Cerinic, M. Clinical, morphological features and prognostic factors associated with interstitial lung disease in primary Sjögren’s syndrome: A systematic review from the Italian Society of Rheumatology. Autoimmun. Rev. 2020, 19, 103447. [Google Scholar] [CrossRef]
- Betteridge, Z.; Tansley, S.; Shaddick, G.; Chinoy, H.; Cooper, R.G.; New, R.P.; Lilleker, J.B.; Vencovsky, J.; Chazarain, L.; Danko, K.; et al. Frequency, mutual exclusivity and clinical association of myositis autoantibodies in a combined European cohort of idiopathic inflammatory myopathies patients. J. Autoimmun. 2019, 101, 48–55. [Google Scholar] [CrossRef]
- Slight-Webb, S.; Lu, R.; Ritterhouse, L.L.; Munroe, M.E.; Maecker, H.H.T.; Fathman, C.G.; Utz, P.J.; Merrill, J.T.; Guthridge, J.M.; James, J.A. Autoantibody-positive Healthy individuals display unique immune profiles that may regulate autoimmunity. Arthritis Rheumatol. 2016, 68, 2492–2502. [Google Scholar] [CrossRef]
- Maheswaranatha, M.; Johannemann, A.; Weiner, J.J.; Jessee, R.; Eudy, A.M.; Criscione-Schreiber, L. Real world utilization of the myositis autoantibody panel. Clin. Rheumatol. 2021, 40, 3195–3205. [Google Scholar] [CrossRef]
- Cavazzana, I.; Fredi, M.; Ceribelli, A.; Mordenti, C.; Ferrari, F.; Carabellese, N.; Tincani, A.; Satoh, M.; Franceschini, F. Testing for myositis specific autoantibodies: Comparison between line blot and immunoprecipitation assays in 57 myositis sera. J. Immunol. Methods 2016, 433, 1–5. [Google Scholar] [CrossRef]
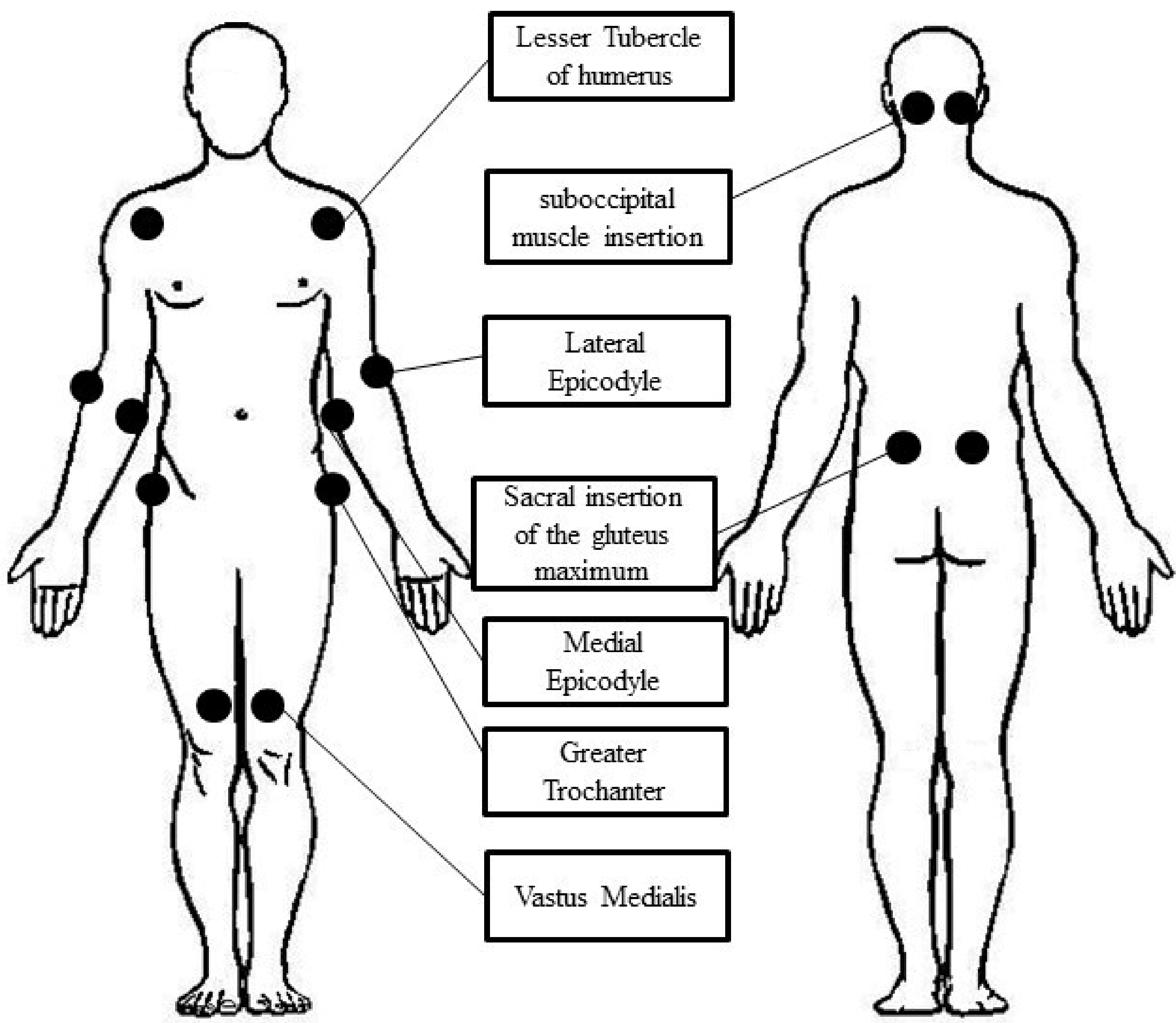

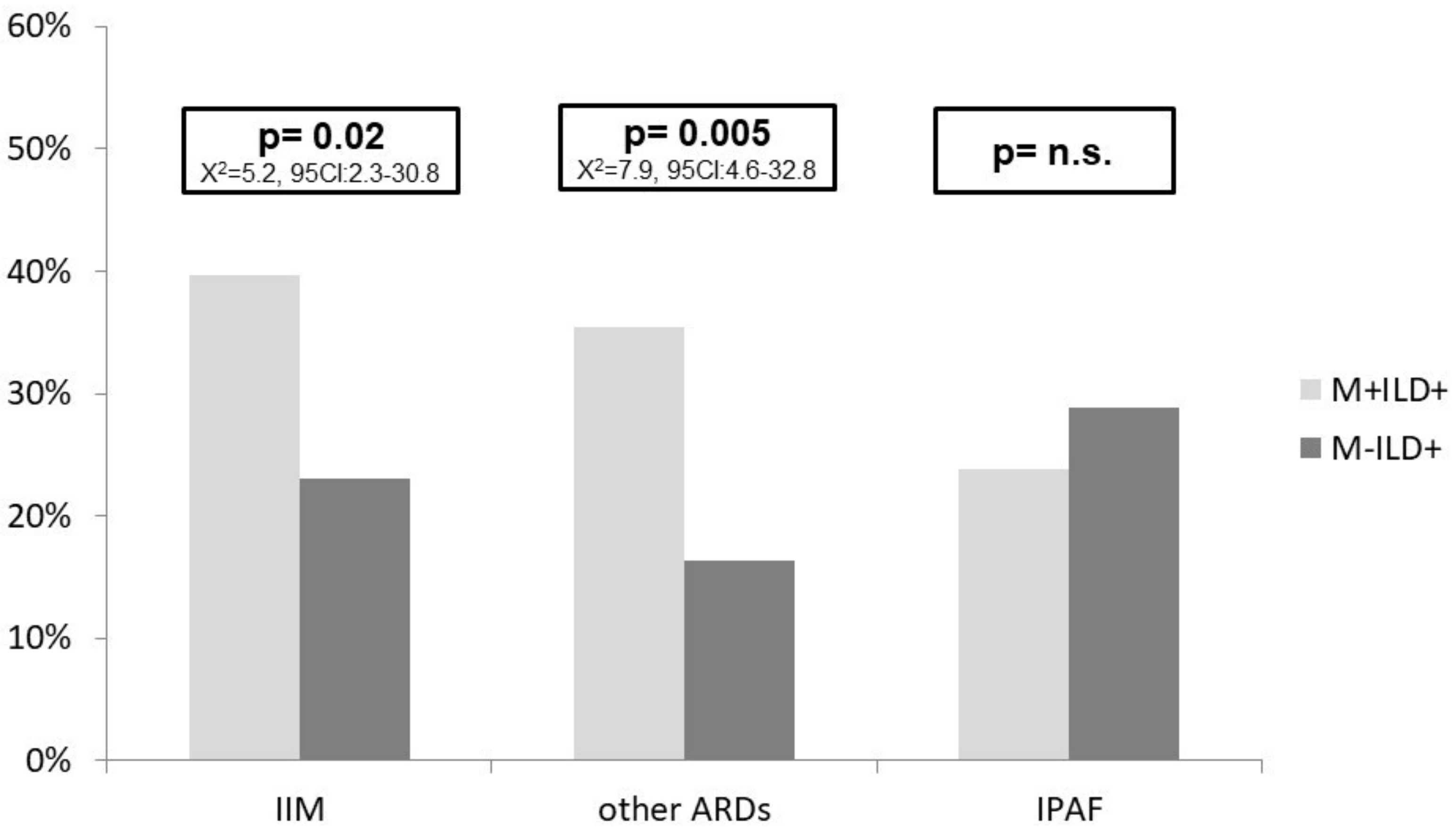

{kind=link}
{kind=link}
{kind=link}
| Item | M+ILD+ (n = 63) | M-ILD+ (n = 104) | p |
|---|---|---|---|
| Age | 62.9 ± 10.6 | 64.3 ± 10.5 | n.s. |
| Female | 90.5% | 52.9% | <0.0001 X2 = 25 95CI:24.1–48.4 |
| Arthritis/PMR | 22.2% | 34.6% | n.s. |
| RP | 49.2% | 30.8% | 0.02 X2= 5.6 95CI: 3.2–32.9 |
| Sclerodattilia/puffy fingers | 17.5% | 12.5% | n.s. |
| Telangiectasias | 14.3% | 8.7% | n.s. |
| Skin rashes # | 17.5% | 9.6% | n.s. |
| Gottron’s papules/sign, heliotrope rash | 11.1% | 3.8% | n.s. |
| Unexplained fever | 30.1% | 25% | n.s. |
| Sicca syndrome | 34.9% | 25% | n.s. |
| All increased seric muscle enzymes | 22.2% | 29.8% | n.s. |
| Increased CPK and/or LDH | 19% | 29.8% | n.s. |
| Dysphagia | 44.4% | 38.5% | n.s. |
| NVC+ | 20.6% | 21.2% | n.s. |
| Bushy capillaries in NVC | 27% | 37.5% | n.s. |
| Exocrine gland functional tests § | 19% | 16.3% | n.s. |
| History of cancer | 14.3% | 6.7% | n.s. |
| Accompanying features of IIMs * | 3.9 ± 2 | 3.6 ± 1.9 | n.s. |
| At least 1 feature specific for IIM ° | 44.4% | 38.5% | n.s. |
| Respiratory features | |||
| Dispnoea | 92.1% | 90.4% | n.s. |
| Cough | 49.2% | 48.1% | n.s. |
| FVC% | 87 ± 25.6 | 86 ± 23.2 | n.s. |
| DLCO% | 65 ± 20.4 | 63.7 ± 20.1 | n.s. |
| Oxygen support | 36.5% | 34.6% | n.s. |
| Radiologic pattern of ILD | |||
| NSIP | 61.9% | 48.1% | n.s. |
| OP | 11.1% | 17.3% | n.s. |
| UIP-like | 27% | 37.5% | n.s. |
| Combined | 6.4% | 6.7% | n.s. |
| Indeterminate | 4.7% | 3.8% | n.s. |
| Dependent Variable: Positivity for MSA | ||
|---|---|---|
| Item | p | OR (95%CI) |
| Myalgia | 0.02 | 2.47 (1.2–5.3) |
| Typical skin rashes * | 0.06 | 3.6 (0.96–14) |
| Proximal weakness | 0.03 | 2.6 (1.1–6) |
| Dysphagia | 0.31 | 1.5 (0.7–3.2) |
| Dependent Variable: Positivity for MSA/MAA | ||
| Item | p | OR (95%CI) |
| Myalgia | 0.04 | 2.1 (1–4.1) |
| Raynaud’s phenomenon | 0.04 | 2.1 (1–4.4) |
| Puffy fingers | 0.22 | 2.2 (0.6–8.4) |
| Telangiectasia | 0.93 | 0.93 (0.2–4.4) |
| Typical skin rashes * | 0.35 | 2.2 (0.4–11.9) |
| Proximal weakness | 0.18 | 1.7 (0.8–4) |
| Dysphagia | 0.29 | 1.5 (0.7–3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sambataro, G.; Ferrara, C.A.; Spadaro, C.; Torrisi, S.E.; Vignigni, G.; Vancheri, A.; Muscato, G.; Del Papa, N.; Colaci, M.; Malatino, L.; et al. A New Method for the Assessment of Myalgia in Interstitial Lung Disease: Association with Positivity for Myositis-Specific and Myositis-Associated Antibodies. Diagnostics 2022, 12, 1139. https://doi.org/10.3390/diagnostics12051139
Sambataro G, Ferrara CA, Spadaro C, Torrisi SE, Vignigni G, Vancheri A, Muscato G, Del Papa N, Colaci M, Malatino L, et al. A New Method for the Assessment of Myalgia in Interstitial Lung Disease: Association with Positivity for Myositis-Specific and Myositis-Associated Antibodies. Diagnostics. 2022; 12(5):1139. https://doi.org/10.3390/diagnostics12051139
Chicago/Turabian StyleSambataro, Gianluca, Chiara Alfia Ferrara, Carla Spadaro, Sebastiano Emanuele Torrisi, Giovanna Vignigni, Ada Vancheri, Giuseppe Muscato, Nicoletta Del Papa, Michele Colaci, Lorenzo Malatino, and et al. 2022. "A New Method for the Assessment of Myalgia in Interstitial Lung Disease: Association with Positivity for Myositis-Specific and Myositis-Associated Antibodies" Diagnostics 12, no. 5: 1139. https://doi.org/10.3390/diagnostics12051139
APA StyleSambataro, G., Ferrara, C. A., Spadaro, C., Torrisi, S. E., Vignigni, G., Vancheri, A., Muscato, G., Del Papa, N., Colaci, M., Malatino, L., Palmucci, S., Cavagna, L., Zanframundo, G., Ferro, F., Baldini, C., Sambataro, D., & Vancheri, C. (2022). A New Method for the Assessment of Myalgia in Interstitial Lung Disease: Association with Positivity for Myositis-Specific and Myositis-Associated Antibodies. Diagnostics, 12(5), 1139. https://doi.org/10.3390/diagnostics12051139