
Biliary Atresia: A Complex Hepatobiliary Disease with Variable Gene Involvement, Diagnostic Procedures, and Prognosis



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Histology of the Developing Fetal Liver
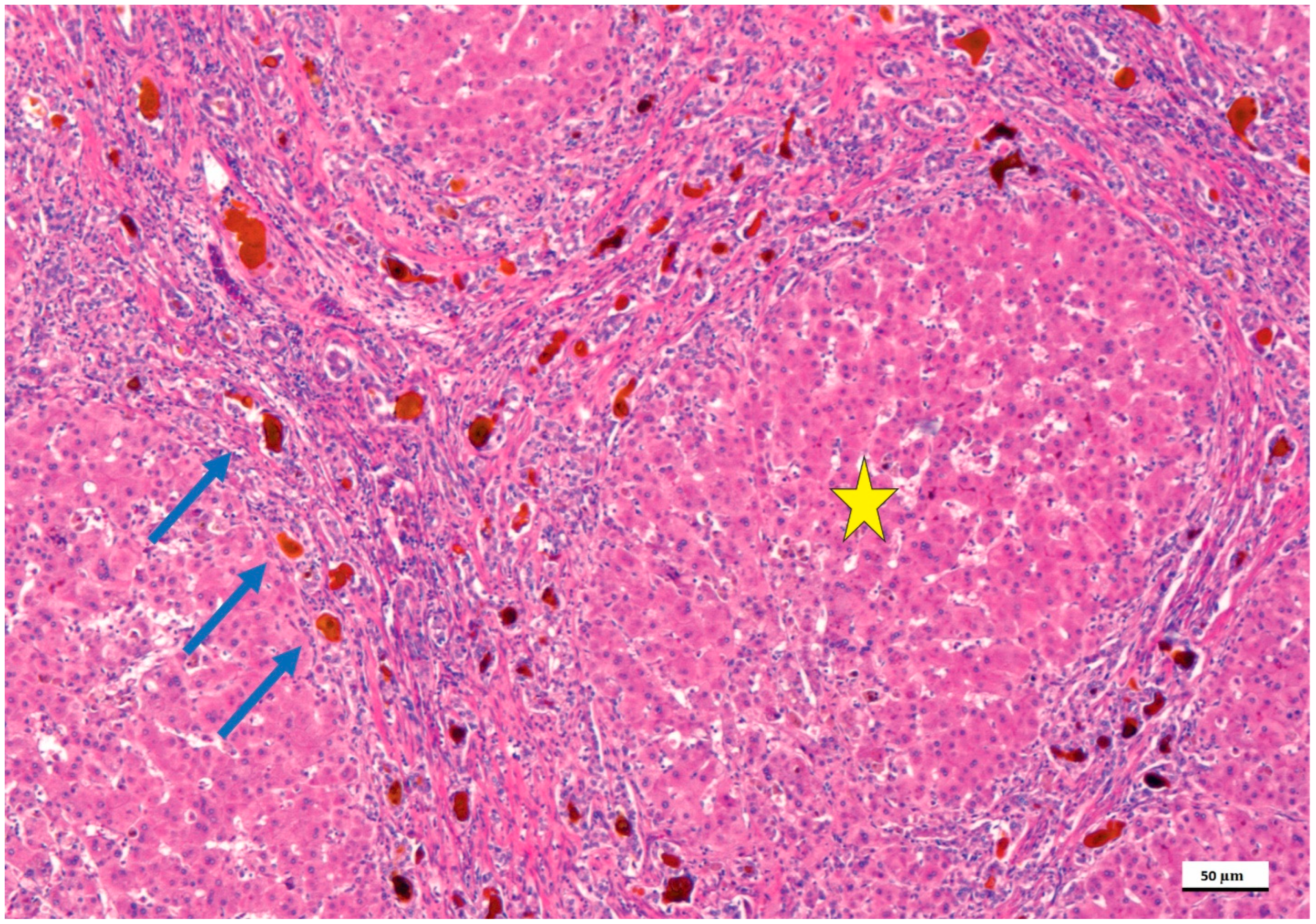
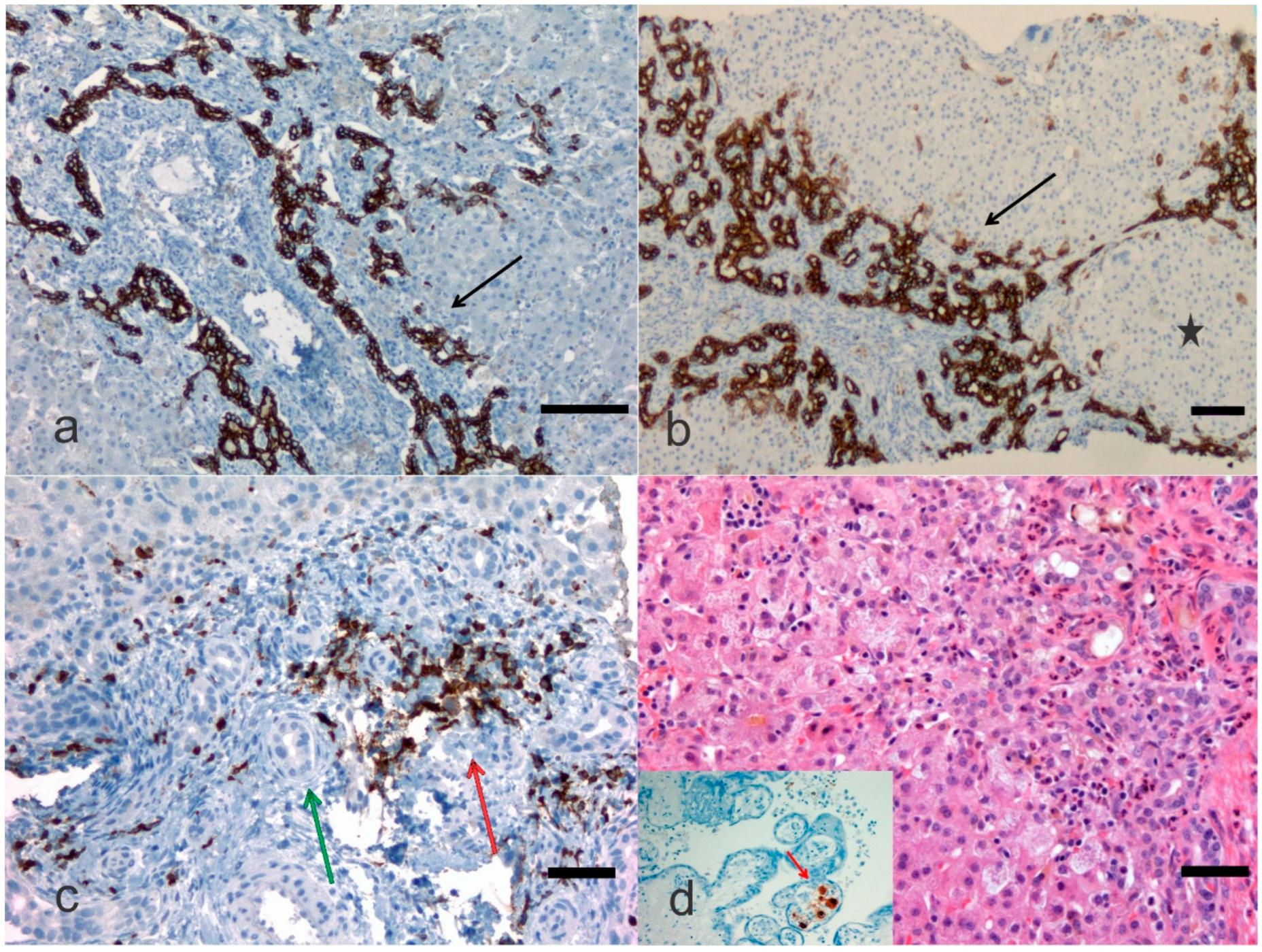
4. Pathological Anatomy of the Biliary Atresia
5. Genetic Complexities and Perspectives
6. Non-Genetic Ambiguities
7. Treatment
8. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ananth, R. Neonatal Cholestasis: A Primer of Selected Etiologies. Pediatr. Ann. 2018, 47, e433–e439. [Google Scholar] [CrossRef]
- Pang, W.B.; Zhang, T.C.; Chen, Y.J.; Peng, C.H.; Wang, Z.M.; Wu, D.Y.; Wang, K. Ten-Year Experience in the Prevention of Post-Kasai Cholangitis. Surg. Infect. 2019, 20, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.W.E.; Lee, K.H.; Wong, H.Y.V.; Tsui, S.Y.B.; Mou, J.W.C.; Tam, Y.H.P. Ten-Year Native Liver Survival Rate After Laparoscopic and Open Kasai Portoenterostomy for Biliary Atresia. J. Laparoendosc. Adv. Surg. Tech. A 2019, 29, 121–125. [Google Scholar] [CrossRef]
- Wong, Z.H.; Davenport, M. What Happens after Kasai for Biliary Atresia? A European Multicenter Survey. Eur. J. Pediatr. Surg. 2019, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.V.; Cowles, R.A.; Kato, T.; Hardy, M.A. Morio Kasai: A remarkable impact beyond the Kasai procedure. J. Pediatr. Surg. 2012, 47, 1023–1027. [Google Scholar] [CrossRef]
- Sergi, C.M. Genetics of Biliary Atresia: A Work in Progress for a Disease with an Unavoidable Sequela into Liver Cirrhosis following Failure of Hepatic Portoenterostomy. In Liver Cirrhosis—Debates and Current Challenges; Tsoulfas, G., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Sergi, C.; Benstz, J.; Feist, D.; Nutzenadel, W.; Otto, H.F.; Hofmann, W.J. Bile duct to portal space ratio and ductal plate remnants in liver disease of infants aged less than 1 year. Pathology 2008, 40, 260–267. [Google Scholar] [CrossRef]
- Obayashi, J.; Tanaka, K.; Ohyama, K.; Manabe, S.; Nagae, H.; Shima, H.; Sato, H.; Furuta, S.; Wakisaka, M.; Koike, J.; et al. Relation between amount of bile ducts in portal canal and outcomes in biliary atresia. Pediatr. Surg. Int. 2016, 32, 833–838. [Google Scholar] [CrossRef]
- Chen, G.; Xue, P.; Zheng, S.; Chen, L.; Ma, Y. A pathological scoring system in the diagnosis and judgment of prognosis of biliary atresia. J. Pediatr. Surg. 2015, 50, 2119–2123. [Google Scholar] [CrossRef]
- Czubkowski, P.; Cielecka-Kuszyk, J.; Rurarz, M.; Kaminska, D.; Markiewicz-Kijewska, M.; Pawlowska, J. The limited prognostic value of liver histology in children with biliary atresia. Ann. Hepatol. 2015, 14, 902–909. [Google Scholar] [CrossRef]
- Safwan, M.; Ramachandran, P.; Vij, M.; Shanmugam, N.; Rela, M. Impact of ductal plate malformation on survival with native liver in children with biliary atresia. Pediatr. Surg. Int. 2015, 31, 837–843. [Google Scholar] [CrossRef]
- Vukovic, J.; Grizelj, R.; Bojanic, K.; Coric, M.; Luetic, T.; Batinica, S.; Kujundzic-Tiljak, M.; Schroeder, D.R.; Sprung, J. Ductal plate malformation in patients with biliary atresia. Eur. J. Pediatr. 2012, 171, 1799–1804. [Google Scholar] [CrossRef]
- Yamaguti, D.C.; Patricio, F.R. Morphometrical and immunohistochemical study of intrahepatic bile ducts in biliary atresia. Eur. J. Gastroenterol. Hepatol. 2011, 23, 759–765. [Google Scholar] [CrossRef]
- Dorn, L.; Menezes, L.F.; Mikuz, G.; Otto, H.F.; Onuchic, L.F.; Sergi, C. Immunohistochemical detection of polyductin and co-localization with liver progenitor cell markers during normal and abnormal development of the intrahepatic biliary system and in adult hepatobiliary carcinomas. J. Cell. Mol. Med. 2009, 13, 1279–1290. [Google Scholar] [CrossRef][Green Version]
- Pacheco, M.C.; Campbell, K.M.; Bove, K.E. Ductal plate malformation-like arrays in early explants after a Kasai procedure are independent of splenic malformation complex (heterotaxy). Pediatr. Dev. Pathol. 2009, 12, 355–360. [Google Scholar] [CrossRef]
- Shimadera, S.; Iwai, N.; Deguchi, E.; Kimura, O.; Ono, S.; Fumino, S.; Higuchi, K. Significance of ductal plate malformation in the postoperative clinical course of biliary atresia. J. Pediatr. Surg. 2008, 43, 304–307. [Google Scholar] [CrossRef]
- Beiler, H.A.; Sergi, C.; Wagner, G.; Zachariou, Z. Accessory liver in an infant with congenital diaphragmatic hernia. J. Pediatr. Surg. 2001, 36, 1–3. [Google Scholar] [CrossRef]
- Sergi, C.M. Pathology of Childhood and Adolescence. An Illustrated Guide; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Sergi, C.; Schulze, B.R.; Hager, H.D.; Beedgen, B.; Zilow, E.; Linderkamp, O.; Otto, H.F.; Tariverdian, G. Wolf-Hirschhorn syndrome: Case report and review of the chromosomal aberrations associated with diaphragmatic defects. Pathologica 1998, 90, 285–293. [Google Scholar]
- Tremblay, K.D.; Zaret, K.S. Distinct populations of endoderm cells converge to generate the embryonic liver bud and ventral foregut tissues. Dev. Biol. 2005, 280, 87–99. [Google Scholar] [CrossRef]
- Amella, C.; Cappello, F.; Kahl, P.; Fritsch, H.; Lozanoff, S.; Sergi, C. Spatial and temporal dynamics of innervation during the development of fetal human pancreas. Neuroscience 2008, 154, 1477–1487. [Google Scholar] [CrossRef]
- Sergi, C.; Adam, S.; Kahl, P.; Otto, H.F. The remodeling of the primitive human biliary system. Early Hum. Dev. 2000, 58, 167–178. [Google Scholar] [CrossRef]
- Lemaigre, F.P. Development of the biliary tract. Mech. Dev. 2003, 120, 81–87. [Google Scholar] [CrossRef]
- Johnson, C.A.; Gissen, P.; Sergi, C. Molecular pathology and genetics of congenital hepatorenal fibrocystic syndromes. J. Med. Genet. 2003, 40, 311–319. [Google Scholar] [CrossRef]
- Sergi, C.; Kahl, P.; Otto, H.F. Contribution of apoptosis and apoptosis-related proteins to the malformation of the primitive intrahepatic biliary system in Meckel syndrome. Am. J. Pathol. 2000, 156, 1589–1598. [Google Scholar] [CrossRef]
- Carvalho, N.M.N.; Torres, S.M.; Cavalcante, J.C.B.; Ximenes, A.C.M.; Landim Junior, J.A.; Moreira, S. Hepatoportoenterostomy Surgery Technique. J. Pediatr. Surg. 2018, 54, 1715–1718. [Google Scholar] [CrossRef]
- Sergi, C.; Adam, S.; Kahl, P.; Otto, H.F. Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr. Dev. Pathol. 2000, 3, 568–583. [Google Scholar] [CrossRef]
- Russo, P.; Magee, J.C.; Anders, R.A.; Bove, K.E.; Chung, C.; Cummings, O.W.; Finegold, M.J.; Finn, L.S.; Kim, G.E.; Lovell, M.A.; et al. Key Histopathologic Features of Liver Biopsies That Distinguish Biliary Atresia From Other Causes of Infantile Cholestasis and Their Correlation With Outcome: A Multicenter Study. Am. J. Surg. Pathol. 2016, 40, 1601–1615. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, X.; Zhao, J.; Fu, M.; Li, Y.; Zhong, W.; Xia, H.; Zhang, Y.; Zhang, R.Z. The intragenic epistatic association of ADD3 with biliary atresia in Southern Han Chinese population. Biosci. Rep. 2018, 38, BSR20171688. [Google Scholar] [CrossRef]
- Ke, J.; Zeng, S.; Mao, J.; Wang, J.; Lou, J.; Li, J.; Chen, X.; Liu, C.; Huang, L.M.; Wang, B.; et al. Common genetic variants of GPC1 gene reduce risk of biliary atresia in a Chinese population. J. Pediatr. Surg. 2016, 51, 1661–1664. [Google Scholar] [CrossRef]
- Smith, K. Biliary tract: GPC1 genetic risk further links Hedgehog signalling with pathogenesis of biliary atresia. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 127. [Google Scholar] [CrossRef]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef]
- Tian, L.; Ye, Z.; Kafka, K.; Stewart, D.; Anders, R.; Schwarz, K.B.; Jang, Y.Y. Biliary Atresia Relevant Human Induced Pluripotent Stem Cells Recapitulate Key Disease Features in a Dish. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 56–63. [Google Scholar] [CrossRef]
- Zeng, S.; Sun, P.; Chen, Z.; Mao, J.; Wang, J.; Wang, B.; Liu, L. Association between single nucleotide polymorphisms in the ADD3 gene and susceptibility to biliary atresia. PLoS ONE 2014, 9, e107977. [Google Scholar] [CrossRef]
- Li, J.; Gao, W.; Zuo, W.; Liu, X. Association between rs17095355 polymorphism on 10q24 and susceptibility to biliary atresia: A meta-analysis. J. Matern. Fetal Neonatal Med. 2017, 30, 1882–1886. [Google Scholar] [CrossRef]
- Mezina, A.; Karpen, S.J. Genetic contributors and modifiers of biliary atresia. Dig. Dis. 2015, 33, 408–414. [Google Scholar] [CrossRef]
- Choi, W.T.; Kakar, S. Immunohistochemistry in the Diagnosis of Hepatocellular Carcinoma. Gastroenterol. Clin. N. Am. 2017, 46, 311–325. [Google Scholar] [CrossRef]
- Sangkhathat, S.; Laochareonsuk, W.; Maneechay, W.; Kayasut, K.; Chiengkriwate, P. Variants Associated with Infantile Cholestatic Syndromes Detected in Extrahepatic Biliary Atresia by Whole Exome Studies: A 20-Case Series from Thailand. J. Pediatr. Genet. 2018, 7, 67–73. [Google Scholar] [CrossRef]
- Schon, P.; Tsuchiya, K.; Lenoir, D.; Mochizuki, T.; Guichard, C.; Takai, S.; Maiti, A.K.; Nihei, H.; Weil, J.; Yokoyama, T.; et al. Identification, genomic organization, chromosomal mapping and mutation analysis of the human INV gene, the ortholog of a murine gene implicated in left-right axis development and biliary atresia. Hum. Genet. 2002, 110, 157–165. [Google Scholar] [CrossRef]
- Tumgor, G.; Cogulu, O.; Onay, H.; Ekmekci, A.Y.; Aydogdu, S.; Durmaz, B.; Kilic, M.; Ozkinay, F. Unusual presentation of biliary atresia splenic malformation syndrome with autosomal dominant hypospadias. Genet. Couns. 2011, 22, 347–351. [Google Scholar]
- Santos, J.L.; Carvalho, E.; Bezerra, J.A. Advances in biliary atresia: From patient care to research. Braz. J. Med. Biol. Res. 2010, 43, 522–527. [Google Scholar] [CrossRef]
- Davit-Spraul, A.; Baussan, C.; Hermeziu, B.; Bernard, O.; Jacquemin, E. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J. Pediatr. Gastroenterol. Nutr. 2008, 46, 111–112. [Google Scholar] [CrossRef]
- Eminoglu, T.F.; Polat, E.; Gokce, S.; Ezgu, F.S.; Senel, S.; Apaydin, S. Cystic fibrosis presenting with neonatal cholestasis simulating biliary atresia in a patient with a novel mutation. Indian J. Pediatr. 2013, 80, 502–504. [Google Scholar] [CrossRef]
- Demeilliers, C.; Jacquemin, E.; Barbu, V.; Mergey, M.; Paye, F.; Fouassier, L.; Chignard, N.; Housset, C.; Lomri, N.E. Altered hepatobiliary gene expressions in PFIC1: ATP8B1 gene defect is associated with CFTR downregulation. Hepatology 2006, 43, 1125–1134. [Google Scholar] [CrossRef]
- Mao, Y.; Tang, S.; Yang, L.; Li, K. Inhibition of the Notch Signaling Pathway Reduces the Differentiation of Hepatic Progenitor Cells into Cholangiocytes in Biliary Atresia. Cell. Physiol. Biochem. 2018, 49, 1074–1082. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Dong, C.; Feng, J.X.; Huang, Z.H. Clinical Features and Genetic Analysis of Pediatric Patients with Alagille Syndrome Presenting Initially with Liver Function Abnormalities. Curr. Med. Sci. 2018, 38, 304–309. [Google Scholar] [CrossRef]
- Ohashi, K.; Togawa, T.; Sugiura, T.; Ito, K.; Endo, T.; Aoyama, K.; Negishi, Y.; Kudo, T.; Ito, R.; Saitoh, S. Combined genetic analyses can achieve efficient diagnostic yields for subjects with Alagille syndrome and incomplete Alagille syndrome. Acta Paediatr. 2017, 106, 1817–1824. [Google Scholar] [CrossRef]
- Zagory, J.A.; Dietz, W.; Park, A.; Fenlon, M.; Xu, J.; Utley, S.; Mavila, N.; Wang, K.S. Notch signaling promotes ductular reactions in biliary atresia. J. Surg. Res. 2017, 215, 250–256. [Google Scholar] [CrossRef]
- Kohsaka, T.; Yuan, Z.R.; Guo, S.X.; Tagawa, M.; Nakamura, A.; Nakano, M.; Kawasasaki, H.; Inomata, Y.; Tanaka, K.; Miyauchi, J. The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology 2002, 36, 904–912. [Google Scholar] [CrossRef]
- Yang, Y.; Jin, Z.; Dong, R.; Zheng, C.; Huang, Y.; Zheng, Y.; Shen, Z.; Chen, G.; Luo, X.; Zheng, S. MicroRNA-29b/142-5p contribute to the pathogenesis of biliary atresia by regulating the IFN-γ gene. Cell. Death Dis. 2018, 9, 545. [Google Scholar] [CrossRef]
- Hsu, Y.A.; Lin, C.H.; Lin, H.J.; Huang, C.C.; Lin, H.C.; Chen, Y.C.; Chang, C.Y.; Huang, S.H.; Lin, J.M.; Lee, K.R.; et al. Effect of microRNA-155 on the interferon-γ signaling pathway in biliary atresia. Chin. J. Physiol. 2016, 59, 315–322. [Google Scholar] [CrossRef]
- Shimadera, S.; Iwai, N.; Deguchi, E.; Kimura, O.; Fumino, S.; Yokoyama, T. The inv mouse as an experimental model of biliary atresia. J. Pediatr. Surg. 2007, 42, 1555–1560. [Google Scholar] [CrossRef]
- Sadek, K.H.; Ezzat, S.; Abdel-Aziz, S.A.; Alaraby, H.; Mosbeh, A.; Abdel-Rahman, M.H. Macrophage Migration Inhibitory Factor (MIF) Gene Promotor Polymorphism Is Associated with Increased Fibrosis in Biliary Atresia Patients, but Not with Disease Susceptibility. Ann. Hum. Genet. 2017, 81, 177–183. [Google Scholar] [CrossRef]
- Arikan, C.; Berdeli, A.; Ozgenc, F.; Tumgor, G.; Yagci, R.V.; Aydogdu, S. Positive association of macrophage migration inhibitory factor gene-173G/C polymorphism with biliary atresia. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 77–82. [Google Scholar] [CrossRef]
- Liu, F.; Zeng, J.; Zhu, D.; Zhang, R.; Xu, X.; Wang, M.; Zhang, Y.; Xia, H.; Feng, Z. Association of polymorphism in the VEGFA gene 3′-UTR + 936T/C with susceptibility to biliary atresia in a Southern Chinese Han population. J. Clin. Lab. Anal. 2018, 32, e22342. [Google Scholar] [CrossRef]
- Liu, B.; Wei, J.; Li, M.; Jiang, J.; Zhang, H.; Yang, L.; Wu, H.; Zhou, Q. Association of common genetic variants in VEGFA with biliary atresia susceptibility in Northwestern Han Chinese. Gene 2017, 628, 87–92. [Google Scholar] [CrossRef]
- Edom, P.T.; Meurer, L.; da Silveira, T.R.; Matte, U.; dos Santos, J.L. Immunolocalization of VEGF A and its receptors, VEGFR1 and VEGFR2, in the liver from patients with biliary atresia. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 360–368. [Google Scholar] [CrossRef]
- Higashiyama, H.; Ozawa, A.; Sumitomo, H.; Uemura, M.; Fujino, K.; Igarashi, H.; Imaimatsu, K.; Tsunekawa, N.; Hirate, Y.; Kurohmaru, M.; et al. Embryonic cholecystitis and defective gallbladder contraction in the Sox17-haploinsufficient mouse model of biliary atresia. Development 2017, 144, 1906–1917. [Google Scholar] [CrossRef]
- Waisbourd-Zinman, O.; Koh, H.; Tsai, S.; Lavrut, P.M.; Dang, C.; Zhao, X.; Pack, M.; Cave, J.; Hawes, M.; Koo, K.A.; et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology 2016, 64, 880–893. [Google Scholar] [CrossRef]
- Uemura, M.; Ozawa, A.; Nagata, T.; Kurasawa, K.; Tsunekawa, N.; Nobuhisa, I.; Taga, T.; Hara, K.; Kudo, A.; Kawakami, H.; et al. Sox17 haploinsufficiency results in perinatal biliary atresia and hepatitis in C57BL/6 background mice. Development 2013, 140, 639–648. [Google Scholar] [CrossRef]
- Leung, A.K.C.; Leung, A.A.M.; Wong, A.H.C.; Sergi, C.M.; Kamsites, J.K.M. Giardiasis: An overview. Recent Pat. Inflamm. Allergy Drug Discov. 2019, 13, 134–143. [Google Scholar] [CrossRef]
- Kong, T.; Feulefack, J.; Ruether, K.; Shen, F.; Zheng, W.; Chen, X.Z.; Sergi, C. Ethnic Differences in Genetic Ion Channelopathies Associated with Sudden Cardiac Death: A Systematic Review and Meta-Analysis. Ann. Clin. Lab. Sci. 2017, 47, 481–490. [Google Scholar]
- Kawazoe, A.; Shitara, K. Next-generation sequencing and biomarkers for gastric cancer: What is the future? Ther. Adv. Med. Oncol. 2019, 11, 1758835919848189. [Google Scholar] [CrossRef]
- You, Z.; Wen, J.; Cheng, L.; Ye, H.; Li, B. Screening of targeted genes in extrahepatic bile ducts of mice with experimental biliary atresia. Mol. Med. Rep. 2015, 12, 4326–4331. [Google Scholar] [CrossRef]
- Zerfaoui, M.; Naura, A.S.; Errami, Y.; Hans, C.P.; Rezk, B.M.; Park, J.; Elsegeiny, W.; Kim, H.; Lord, K.; Kim, J.G.; et al. Effects of PARP-1 deficiency on airway inflammatory cell recruitment in response to LPS or TNF: Differential effects on CXCR2 ligands and Duffy Antigen Receptor for Chemokines. J. Leukoc. Biol. 2009, 86, 1385–1392. [Google Scholar] [CrossRef]
- Hirano, Y.; Hirano, F.; Fujii, H.; Makino, I. Fibrates suppress chenodeoxycholic acid-induced RANTES expression through inhibition of NF-kappaB activation. Eur. J. Pharmacol. 2002, 448, 19–26. [Google Scholar] [CrossRef]
- Schukfeh, N.; Al-Gamrah, A.; Petersen, C.; Kuebler, J.F. Detection of hepatotropic viruses has no impact on the prognosis after Kasai procedure. J. Pediatr. Surg. 2012, 47, 1828–1832. [Google Scholar] [CrossRef]
- Rauschenfels, S.; Krassmann, M.; Al-Masri, A.N.; Verhagen, W.; Leonhardt, J.; Kuebler, J.F.; Petersen, C. Incidence of hepatotropic viruses in biliary atresia. Eur. J. Pediatr. 2009, 168, 469–476. [Google Scholar] [CrossRef]
- Landing, B.H. Considerations of the pathogenesis of neonatal hepatitis, biliary atresia and choledochal cyst—The concept of infantile obstructive cholangiopathy. Prog. Pediatr. Surg. 1974, 6, 113–139. [Google Scholar]
- Zhang, S.; Goswami, S.; Ma, J.; Meng, L.; Wang, Y.; Zhu, F.; Zhang, D.; Zheng, S.; Dong, R.; Xiao, X.; et al. CD4+T Cell Subset Profiling in Biliary Atresia Reveals ICOS− Regulatory T Cells as a Favorable Prognostic Factor. Front. Pediatr. 2019, 7, 279. [Google Scholar] [CrossRef]
- Ye, J.; Lai, D.; Cao, D.; Tan, L.; Hu, L.; Zha, H.; Yang, J.; Shu, Q. Altered T-Cell Receptor β-Chain and Lactate Dehydrogenase Are Associated With the Immune Pathogenesis of Biliary Atresia. Front Med. 2021, 8, 778500. [Google Scholar] [CrossRef]
- Zhao, X.; Lorent, K.; Escobar-Zarate, D.; Rajagopalan, R.; Loomes, K.M.; Gillespie, K.; Mesaros, C.; Estrada, M.A.; Blair, I.A.; Winkler, J.D.; et al. Impaired Redox and Protein Homeostasis as Risk Factors and Therapeutic Targets in Toxin-Induced Biliary Atresia. Gastroenterology 2020, 159, 1068–1084.e2. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, J.; Zhan, Y.; Chen, G.; Shen, Z.; Zheng, S.; Dong, R. The synthetic toxin biliatresone causes biliary atresia in mice. Lab. Investig. 2020, 100, 1425–1435. [Google Scholar] [CrossRef]
- Bezerra, J.A.; Wells, R.G.; Mack, C.L.; Karpen, S.J.; Hoofnagle, J.H.; Doo, E.; Sokol, R.J. Biliary Atresia: Clinical and Research Challenges for the 21(st) Century. Hepatology 2018, 68, 1163–1173. [Google Scholar] [CrossRef]
- Davenport, M. Biliary atresia: From Australia to the zebrafish. J. Pediatr. Surg. 2016, 51, 200–205. [Google Scholar] [CrossRef]
- Ningappa, M.; So, J.; Glessner, J.; Ashokkumar, C.; Ranganathan, S.; Min, J.; Higgs, B.W.; Sun, Q.; Haberman, K.; Schmitt, L.; et al. The Role of ARF6 in Biliary Atresia. PLoS ONE 2015, 10, e0138381. [Google Scholar] [CrossRef]
- Huang, Y.H.; Shih, H.H.; Tiao, M.M.; Huang, C.C.; Kuo, K.C.; Huang, F.C.; Yang, Y.L.; Chuang, J.H. Toll-like receptor 7 agonist induces hypoplasia of the biliary system in a neonatal mouse model. J. Microbiol. Immunol. Infect. 2018, 51, 166–173. [Google Scholar] [CrossRef]
- Fu, M.; Lin, Z.; Lin, H.; Tong, Y.; Wang, H.; Chen, H.; Chen, Y.; Zhang, R. A Silver Nanoparticle Method for Ameliorating Biliary Atresia Syndrome in Mice. J. Vis. Exp. 2018, 140, e58158. [Google Scholar] [CrossRef]
- Lakshminarayanan, B.; Davenport, M. Biliary atresia: A comprehensive review. J. Autoimmun. 2016, 73, 1–9. [Google Scholar] [CrossRef]
- Ihn, K.; Na, Y.; Ho, I.G.; Lee, D.; Koh, H.; Han, S.J. A periodic comparison of the survival and prognostic factors of biliary atresia after Kasai portoenterostomy: A single-center study in Korea. Pediatr. Surg. Int. 2019, 35, 285–292. [Google Scholar] [CrossRef]
- Parolini, F.; Boroni, G.; Milianti, S.; Tonegatti, L.; Armellini, A.; Garcia Magne, M.; Pedersini, P.; Torri, F.; Orizio, P.; Benvenuti, S.; et al. Biliary atresia: 20–40-year follow-up with native liver in an Italian centre. J. Pediatr. Surg. 2018, 54, 1440–1444. [Google Scholar] [CrossRef]
- Hasan, M.S.; Karim, A.B.; Rukunuzzaman, M.; Haque, A.; Akhter, M.A.; Shoma, U.K.; Yasmin, F.; Rahman, M.A. Role of Liver Biopsy in the Diagnosis of Neonatal Cholestasis due to Biliary Atresia. Mymensingh Med. J. 2018, 27, 826–833. [Google Scholar]
- Madadi-Sanjani, O.; Kuebler, J.F.; Dippel, S.; Gigina, A.; Falk, C.S.; Vieten, G.; Petersen, C.; Klemann, C. Long-term outcome and necessity of liver transplantation in infants with biliary atresia are independent of cytokine milieu in native liver and serum. Cytokine 2018, 111, 382–388. [Google Scholar] [CrossRef]
- Li, S.; Ma, N.; Meng, X.; Zhang, W.; Sun, C.; Dong, C.; Wang, K.; Wu, B.; Gao, W. The effects of Kasai procedure on living donor liver transplantation for children with biliary atresia. J. Pediatr. Surg. 2018, 54, 1436–1439. [Google Scholar] [CrossRef]
- Witt, M.; van Wessel, D.B.E.; de Kleine, R.H.J.; Bruggink, J.L.M.; Hulscher, J.B.F.; Verkade, H.J.; Netherlands Study Group on Biliary Atresia Registry. Prognosis of Biliary Atresia After 2-year Survival With Native Liver: A Nationwide Cohort Analysis. J. Pediatr. Gastroenterol. Nutr. 2018, 67, 689–694. [Google Scholar] [CrossRef]
- Arafa, R.S.; Abdel Haie, O.M.; El-Azab, D.S.; Abdel-Rahman, A.M.; Sira, M.M. Significant hepatic expression of IL-2 and IL-8 in biliary atresia compared with other neonatal cholestatic disorders. Cytokine 2016, 79, 59–65. [Google Scholar] [CrossRef]
- Santos, J.L.; Kieling, C.O.; Meurer, L.; Vieira, S.; Ferreira, C.T.; Lorentz, A.; Silveira, T.R. The extent of biliary proliferation in liver biopsies from patients with biliary atresia at portoenterostomy is associated with the postoperative prognosis. J. Pediatr. Surg. 2009, 44, 695–701. [Google Scholar] [CrossRef]
- Obayashi, J.; Kawaguchi, K.; Manabe, S.; Nagae, H.; Wakisaka, M.; Koike, J.; Takagi, M.; Kitagawa, H. Prognostic factors indicating survival with native liver after Kasai procedure for biliary atresia. Pediatr. Surg. Int. 2017, 33, 1047–1052. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergi, C.M.; Gilmour, S. Biliary Atresia: A Complex Hepatobiliary Disease with Variable Gene Involvement, Diagnostic Procedures, and Prognosis. Diagnostics 2022, 12, 330. https://doi.org/10.3390/diagnostics12020330
Sergi CM, Gilmour S. Biliary Atresia: A Complex Hepatobiliary Disease with Variable Gene Involvement, Diagnostic Procedures, and Prognosis. Diagnostics. 2022; 12(2):330. https://doi.org/10.3390/diagnostics12020330
Chicago/Turabian StyleSergi, Consolato M., and Susan Gilmour. 2022. "Biliary Atresia: A Complex Hepatobiliary Disease with Variable Gene Involvement, Diagnostic Procedures, and Prognosis" Diagnostics 12, no. 2: 330. https://doi.org/10.3390/diagnostics12020330
APA StyleSergi, C. M., & Gilmour, S. (2022). Biliary Atresia: A Complex Hepatobiliary Disease with Variable Gene Involvement, Diagnostic Procedures, and Prognosis. Diagnostics, 12(2), 330. https://doi.org/10.3390/diagnostics12020330