
α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study

 , , , , ,
, , , , , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA Interference
2.2. Evaluation of PBGD Enzymatic Activity
2.3. Gene Expression Analysis
2.4. Western Blot Analysis
2.5. Statistical Analysis
3. Results
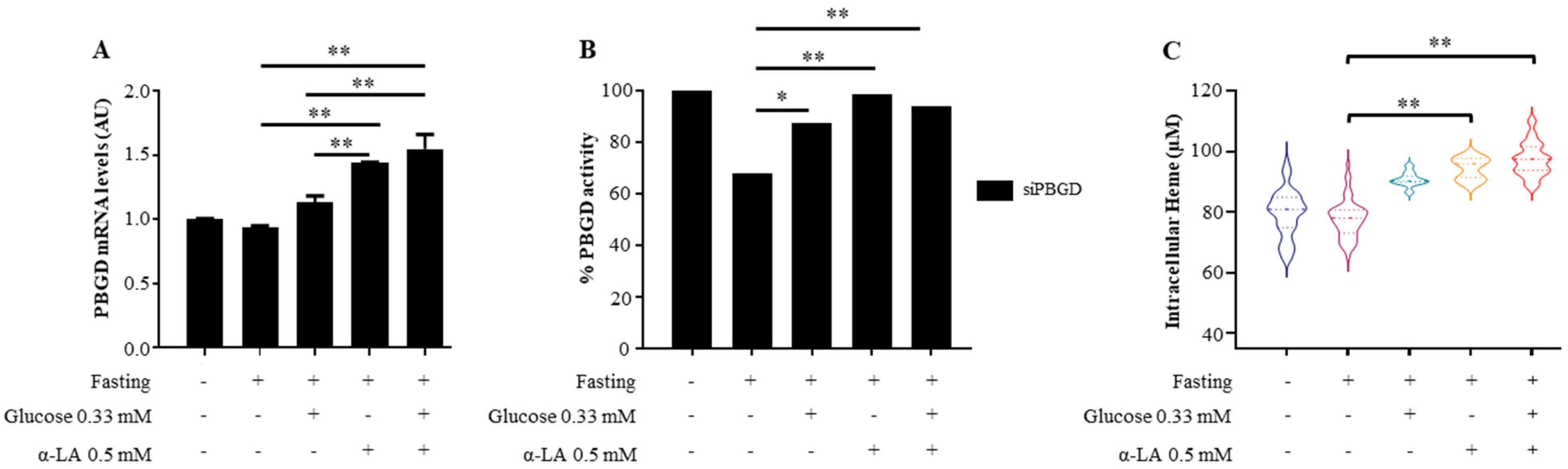
3.1. α-Lipoic Acid Improved Heme Production in siPBGD Cells
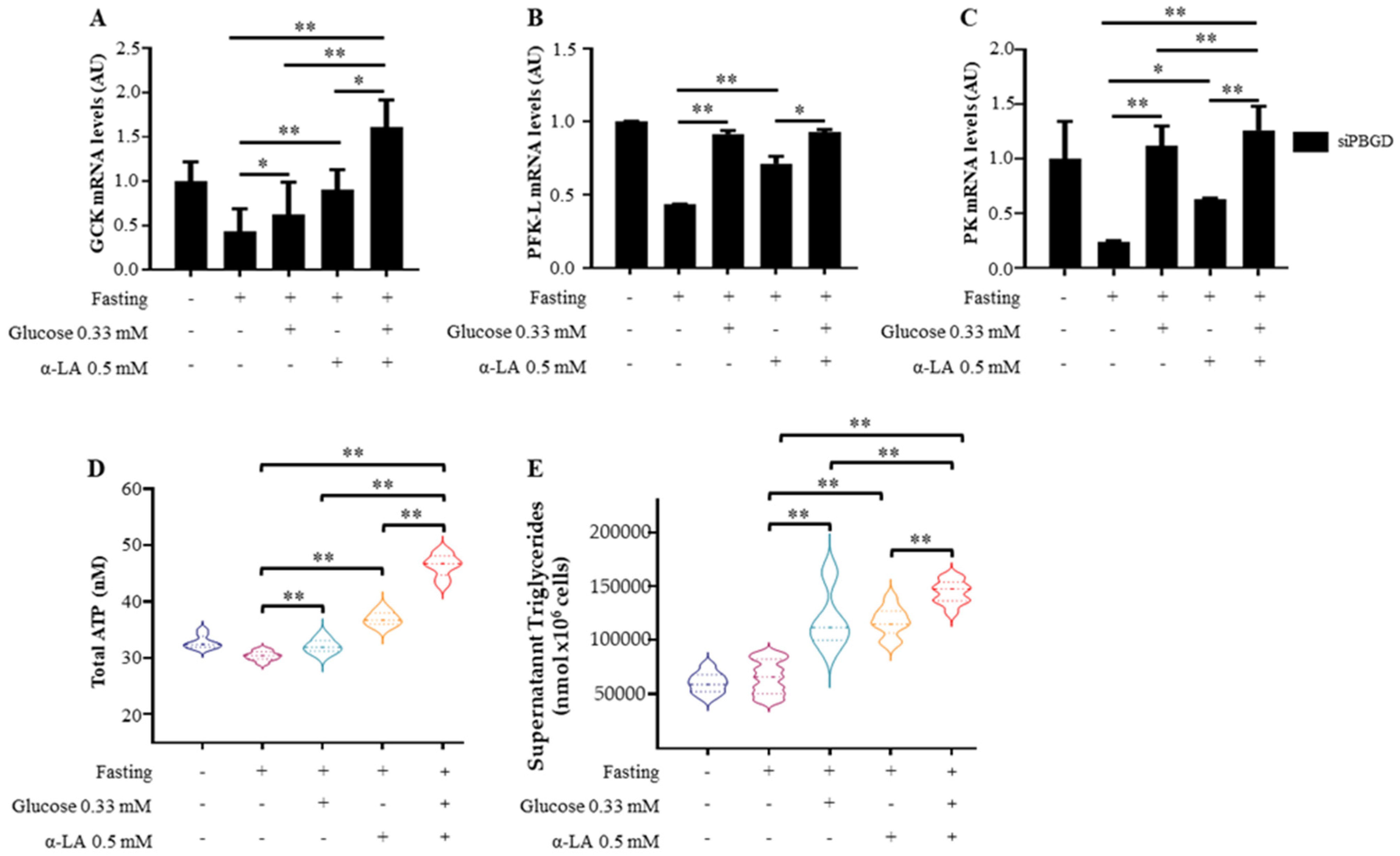
3.2. α-Lipoic Acid Stimulates Glucose Utilization and Provides Energy Supplies during Fasting
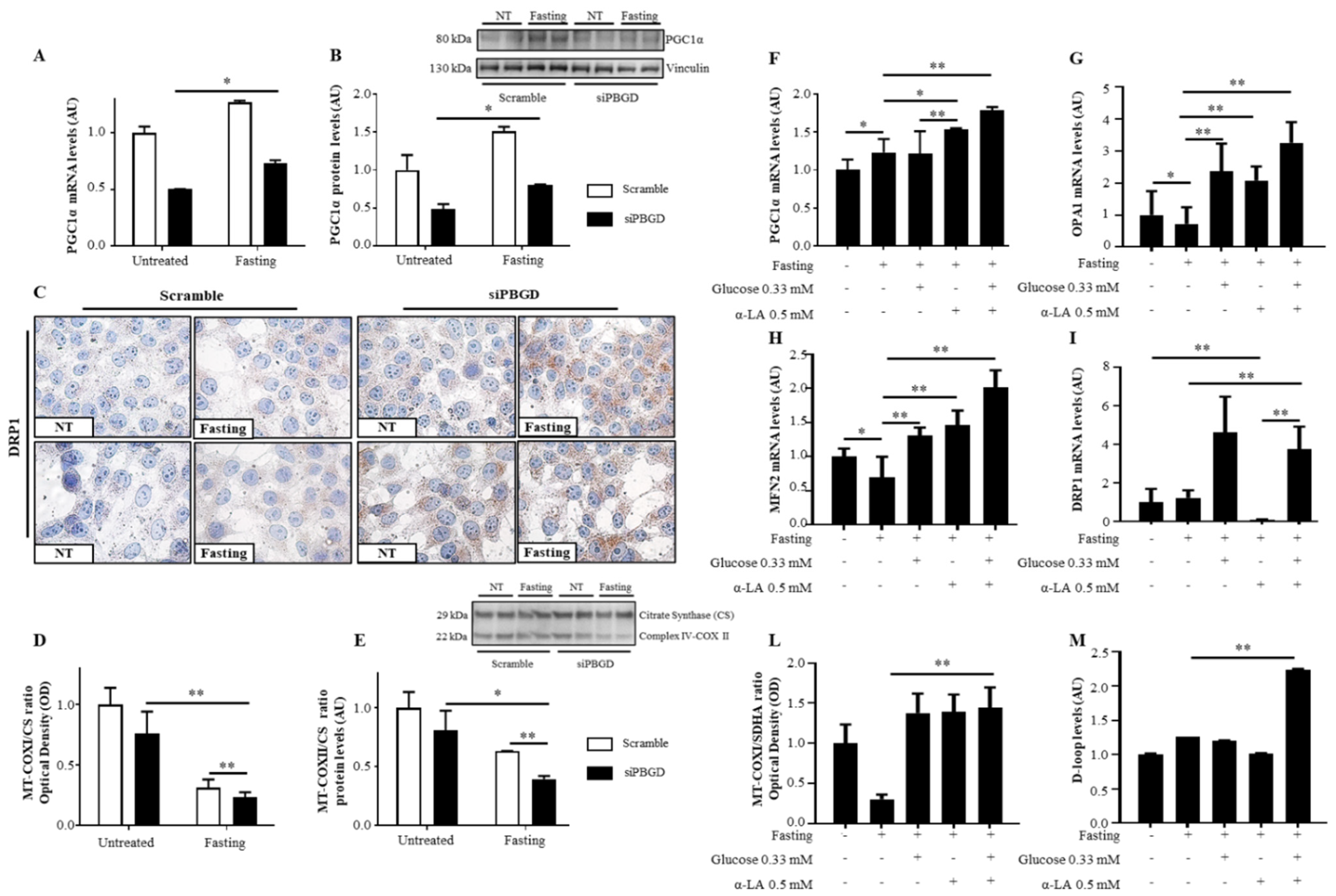
3.3. α-Lipoic Acid Recovered Mitobiogenesis: The Dual Role of PGC1α
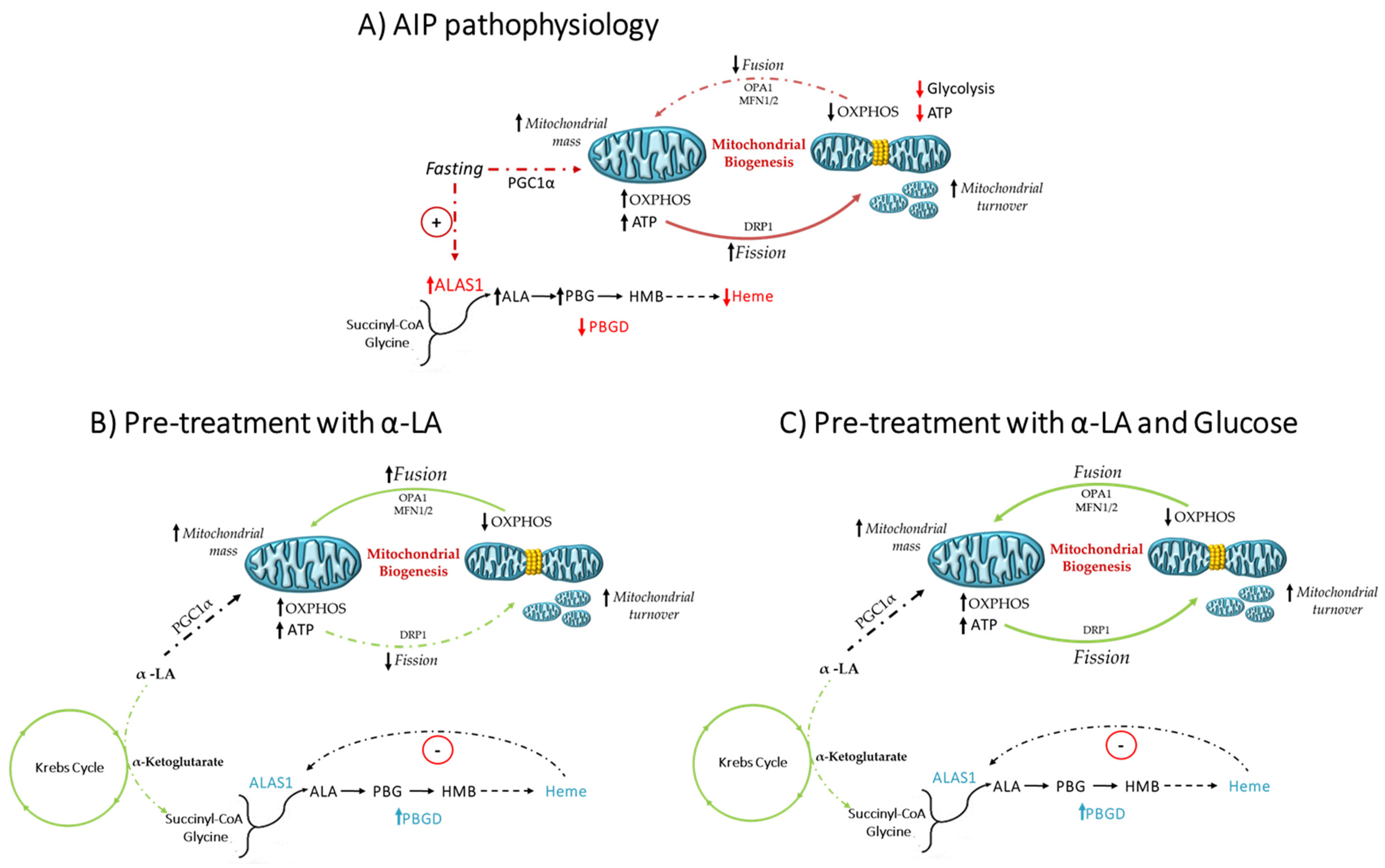
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, B.; Rudnick, S.; Cengia, B.; Bonkovsky, H.L. Acute Hepatic Porphyrias: Review and Recent Progress. Hepatol. Commun. 2019, 3, 193–206. [Google Scholar] [CrossRef]
- Lenglet, H.; Schmitt, C.; Grange, T.; Manceau, H.; Karboul, N.; Bouchet-Crivat, F.; Robreau, A.M.; Nicolas, G.; Lamoril, J.; Simonin, S.; et al. From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Hum. Mol. Genet. 2018, 27, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pischik, E.; Kauppinen, R. An update of clinical management of acute intermittent porphyria. Appl. Clin. Genet. 2015, 8, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, H.P.; Abdalla, D.S.P.; Augusto, O.; Bechara, E.J.H. Free radical generation during δ-Aminolevulinic acid autoxidation: Induction by hemoglobin and connections with porphyrinpathies. Arch. Biochem. Biophys. 1989, 271, 206–216. [Google Scholar] [CrossRef]
- Brennan, M.J.W.; Cantrill, R.C. δ-Aminolaevulinic acid and amino acid neurotransmitters. In The Biological Effects of Glutamic Acid and Its Derivatives; Najjar, V.A., Ed.; Springer: Dordrecht, The Netherlands, 1981; pp. 49–58. [Google Scholar]
- Weiss, T.S.; Pahernik, S.; Scheruebl, I.; Jauch, K.W.; Thasler, W.E. Cellular damage to human hepatocytes through repeated application of 5-aminolevulinic acid. J. Hepatol. 2003, 38, 476–482. [Google Scholar] [CrossRef]
- Chen, B.; Solis-Villa, C.; Hakenberg, J.; Qiao, W.; Srinivasan, R.R.; Yasuda, M.; Balwani, M.; Doheny, D.; Peter, I.; Chen, R.; et al. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease. Hum. Mutat. 2016, 37, 1215–1222. [Google Scholar] [CrossRef]
- Lelli, S.M.; de San Martín Viale, L.C.; Mazzetti, M.B. Response of glucose metabolism enzymes in an acute porphyria model. Role of reactive oxygen species. Toxicology 2005, 216, 49–58. [Google Scholar] [CrossRef]
- Collantes, M.; Serrano-Mendioroz, I.; Benito, M.; Molinet-Dronda, F.; Delgado, M.; Vinaixa, M.; Sampedro, A.; de Enriquez Salamanca, R.; Prieto, E.; Pozo, M.A.; et al. Glucose metabolism during fasting is altered in experimental porphobilinogen deaminase deficiency. Hum. Mol. Genet. 2016, 25, 1318–1327. [Google Scholar] [CrossRef]
- Homedan, C.; Schmitt, C.; Laafi, J.; Gueguen, N.; Desquiret-Dumas, V.; Lenglet, H.; Karim, Z.; Gouya, L.; Deybach, J.-C.; Simard, G.; et al. Mitochondrial energetic defects in muscle and brain of a Hmbs−/− mouse model of acute intermittent porphyria. Hum. Mol. Genet. 2015, 24, 5015–5023. [Google Scholar] [CrossRef]
- Chen, B.; Wang, M.; Gan, L.; Zhang, B.; Desnick, R.J.; Yasuda, M. Characterization of the hepatic transcriptome following phenobarbital induction in mice with AIP. Mol. Genet. Metab. 2019, 128, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Lenglet, H.; Yu, A.; Delaby, C.; Benecke, A.; Lefebvre, T.; Letteron, P.; Paradis, V.; Wahlin, S.; Sandberg, S.; et al. Recurrent attacks of acute hepatic porphyria: Major role of the chronic inflammatory response in the liver. J. Intern. Med. 2018, 284, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Fontanellas, A.; Ávila, M.A.; Anderson, K.E.; Deybach, J.C. Current and innovative emerging therapies for porphyrias with hepatic involvement. J. Hepatol. 2019, 71, 422–433. [Google Scholar] [CrossRef]
- Solares, I.; Izquierdo-Sánchez, L.; Morales-Conejo, M.; Jericó, D.; Castelbón, F.J.; Córdoba, K.M.; Sampedro, A.; Lumbreras, C.; Moreno-Aliaga, M.J.; Enríquez de Salamanca, R.; et al. High Prevalence of Insulin Resistance in Asymptomatic Patients with Acute Intermittent Porphyria and Liver-Targeted Insulin as a Novel Therapeutic Approach. Biomedicines 2021, 9, 255. [Google Scholar] [CrossRef]
- Andersson, C.; Bylesjö, I.; Lithner, F. Effects of diabetes mellitus on patients with acute intermittent porphyria. J. Intern. Med. 1999, 245, 193–197. [Google Scholar] [CrossRef]
- Ko, C.Y.; Lo, Y.M.; Xu, J.H.; Chang, W.C.; Huang, D.W.; Wu, J.S.; Yang, C.H.; Huang, W.C.; Shen, S.C. Alpha-lipoic acid alleviates NAFLD and triglyceride accumulation in liver via modulating hepatic NLRP3 inflammasome activation pathway in type 2 diabetic rats. Food Sci. Nutr. 2021, 9, 2733–2742. [Google Scholar] [CrossRef]
- Rahmanabadi, A.; Mahboob, S.; Amirkhizi, F.; Hosseinpour-Arjmand, S.; Ebrahimi-Mameghani, M. Oral α-lipoic acid supplementation in patients with non-alcoholic fatty liver disease: Effects on adipokines and liver histology features. Food Funct. 2019, 10, 4941–4952. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpour-Arjmand, S.; Amirkhizi, F.; Ebrahimi-Mameghani, M. The effect of alpha-lipoic acid on inflammatory markers and body composition in obese patients with non-alcoholic fatty liver disease: A randomized, double-blind, placebo-controlled trial. J. Clin. Pharm. Ther. 2019, 44, 258–267. [Google Scholar] [CrossRef]
- Hsiao, K.J.; Lee, F.Y.; Wu, S.J.; Chang, W.J. Determination of erythrocyte porphobilinogen deaminase activity using porphobilinogen as substrate. Clin. Chim. Acta Int. J. Clin. Chem. 1987, 168, 257–258. [Google Scholar] [CrossRef]
- Stein, P.E.; Badminton, M.N.; Rees, D.C. Update review of the acute porphyrias. Br. J. Haematol. 2017, 176, 527–538. [Google Scholar] [CrossRef]
- Phillips, J.D. Heme biosynthesis and the porphyrias. Mol. Genet. Metab. 2019, 128, 164–177. [Google Scholar] [CrossRef]
- Storjord, E.; Dahl, J.A.; Landsem, A.; Ludviksen, J.K.; Karlsen, M.B.; Karlsen, B.O.; Brekke, O.-L. Lifestyle factors including diet and biochemical biomarkers in acute intermittent porphyria: Results from a case-control study in northern Norway. Mol. Genet. Metab. 2019, 128, 254–270. [Google Scholar] [CrossRef]
- García-Diz, L.; Murcia, M.A.; Gris, J.L.; Pons, A.; Monteagudo, C.; Martínez-Tomé, M.; Jiménez-Monreal, A.M. Assessing nutritional status of acute intermittent porphyria patients. Eur. J. Clin. Investig. 2012, 42, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Herrick, A.L.; Fisher, B.M.; Moore, M.R.; Cathcart, S.; McColl, K.E.L.; Goldberg, A. Elevation of blood lactate and pyruvate levels in acute intermittent porphyria—A reflection of haem deficiency? Clin. Chim. Acta 1990, 190, 157–162. [Google Scholar] [CrossRef]
- Di Pierro, E.; Granata, F. Nutrients and Porphyria: An Intriguing Crosstalk. Int. J. Mol. Sci. 2020, 21, 3462. [Google Scholar] [CrossRef] [PubMed]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.-C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef]
- Ostrowski, J.; Kostrzewska, E.; Michalak, T.; Zawirska, B.; Medrzejewski, W.; Gregor, A. Abnormalities in liver function and morphology and impaired aminopyrine metabolism in hereditary hepatic porphyrias. Gastroenterology 1983, 85, 1131–1137. [Google Scholar] [CrossRef]
- Berger, S.; Stattmann, M.; Cicvaric, A.; Monje, F.J.; Coiro, P.; Hotka, M.; Ricken, G.; Hainfellner, J.; Greber-Platzer, S.; Yasuda, M.; et al. Severe hydroxymethylbilane synthase deficiency causes depression-like behavior and mitochondrial dysfunction in a mouse model of homozygous dominant acute intermittent porphyria. Acta Neuropathol. Commun. 2020, 8, 38. [Google Scholar] [CrossRef]
- Shetty, T.; Sishtla, K.; Park, B.; Repass, M.J.; Corson, T.W. Heme Synthesis Inhibition Blocks Angiogenesis via Mitochondrial Dysfunction. Iscience 2020, 23, 101391. [Google Scholar] [CrossRef]
- Balwani, M.; Wang, B.; Anderson, K.E.; Bloomer, J.R.; Bissell, D.M.; Bonkovsky, H.L.; Phillips, J.D.; Desnick, R.J. Acute hepatic porphyrias: Recommendations for evaluation and long-term management. Hepatology 2017, 66, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Willandt, B.; Langendonk, J.G.; Biermann, K.; Meersseman, W.; D’Heygere, F.; George, C.; Verslype, C.; Monbaliu, D.; Cassiman, D. Liver Fibrosis Associated with Iron Accumulation Due to Long-Term Heme-Arginate Treatment in Acute Intermittent Porphyria: A Case Series. JIMD Rep. 2016, 25, 77–81. [Google Scholar] [PubMed]
- Storjord, E.; Dahl, J.A.; Landsem, A.; Fure, H.; Ludviksen, J.K.; Goldbeck-Wood, S.; Karlsen, B.O.; Berg, K.S.; Mollnes, T.E.; Nielsen, E.W.; et al. Systemic inflammation in acute intermittent porphyria: A case-control study. Clin. Exp. Immunol. 2017, 187, 466–479. [Google Scholar] [CrossRef][Green Version]
- Solmonson, A.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef]
- Daher, R.; Mansouri, A.; Martelli, A.; Bayart, S.; Manceau, H.; Callebaut, I.; Moulouel, B.; Gouya, L.; Puy, H.; Kannengiesser, C.; et al. GLRX5 mutations impair heme biosynthetic enzymes ALA synthase 2 and ferrochelatase in Human congenital sideroblastic anemia. Mol. Genet. Metab. 2019, 128, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Vilas, G.L.; Aldonatti, C.; San Martín de Viale, L.C.; de Ríos Molina, M.C. Effect of alpha lipoic acid amide on hexachlorobenzene porphyria. Biochem. Mol. Biol. Int. 1999, 47, 815–823. [Google Scholar]
- Székely, E.; Szentmihályi, K.; Tasnádi, G.; Várnai, K.; Blázovics, A. Effect of alpha-lipoic acid on the porphyria cutanea tarda patients with type 2 diabetes mellitus and heavy drinkers. Z. Gastroenterol. 2005, 43, 136. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longo, M.; Paolini, E.; Meroni, M.; Duca, L.; Motta, I.; Fracanzani, A.L.; Di Pierro, E.; Dongiovanni, P. α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study. Diagnostics 2021, 11, 1628. https://doi.org/10.3390/diagnostics11091628
Longo M, Paolini E, Meroni M, Duca L, Motta I, Fracanzani AL, Di Pierro E, Dongiovanni P. α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study. Diagnostics. 2021; 11(9):1628. https://doi.org/10.3390/diagnostics11091628
Chicago/Turabian StyleLongo, Miriam, Erika Paolini, Marica Meroni, Lorena Duca, Irene Motta, Anna Ludovica Fracanzani, Elena Di Pierro, and Paola Dongiovanni. 2021. "α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study" Diagnostics 11, no. 9: 1628. https://doi.org/10.3390/diagnostics11091628
APA StyleLongo, M., Paolini, E., Meroni, M., Duca, L., Motta, I., Fracanzani, A. L., Di Pierro, E., & Dongiovanni, P. (2021). α-Lipoic Acid Improves Hepatic Metabolic Dysfunctions in Acute Intermittent Porphyria: A Proof-of-Concept Study. Diagnostics, 11(9), 1628. https://doi.org/10.3390/diagnostics11091628