
Conserved Sequence Analysis of Influenza A Virus HA Segment and Its Application in Rapid Typing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Influenza Virus Dataset
2.2. Conserved Sequence Searching
2.2.1. Protein Conserved Sequence
2.2.2. Nucleotide Conserved Sequence
2.3. Polymerase Chain Reaction
2.3.1. Influenza Virus Template Plasmid
2.3.2. PCR Primers Design
2.3.3. PCR Experiment
3. Results
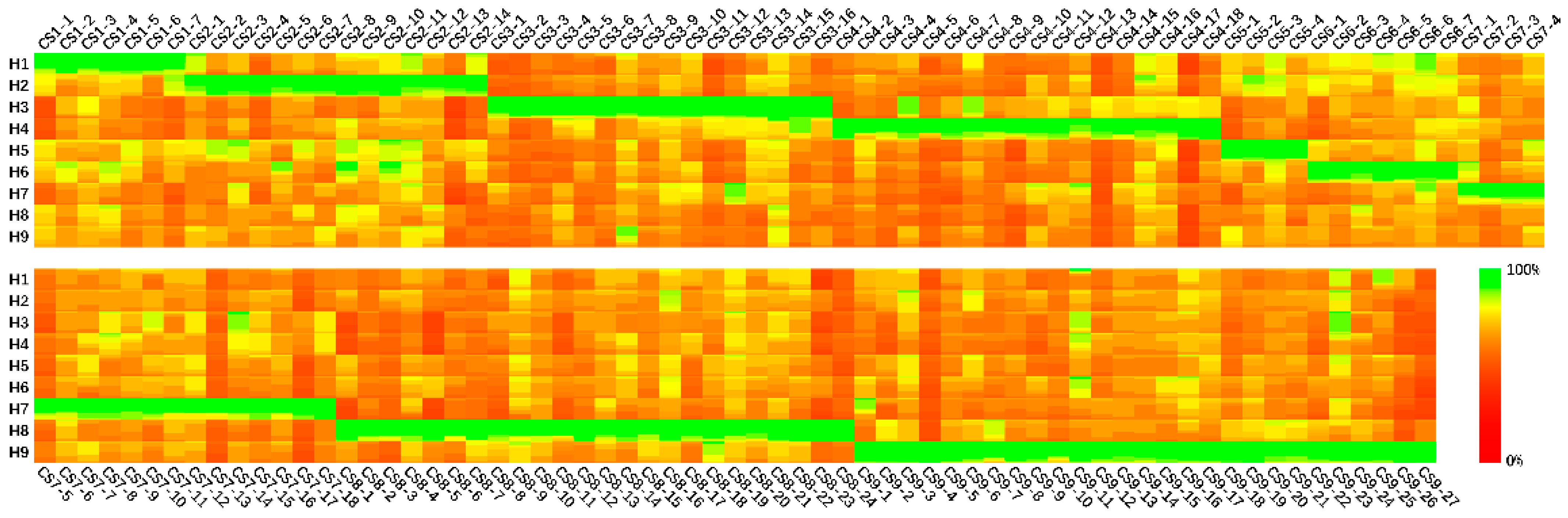
3.1. Conserved Sequences
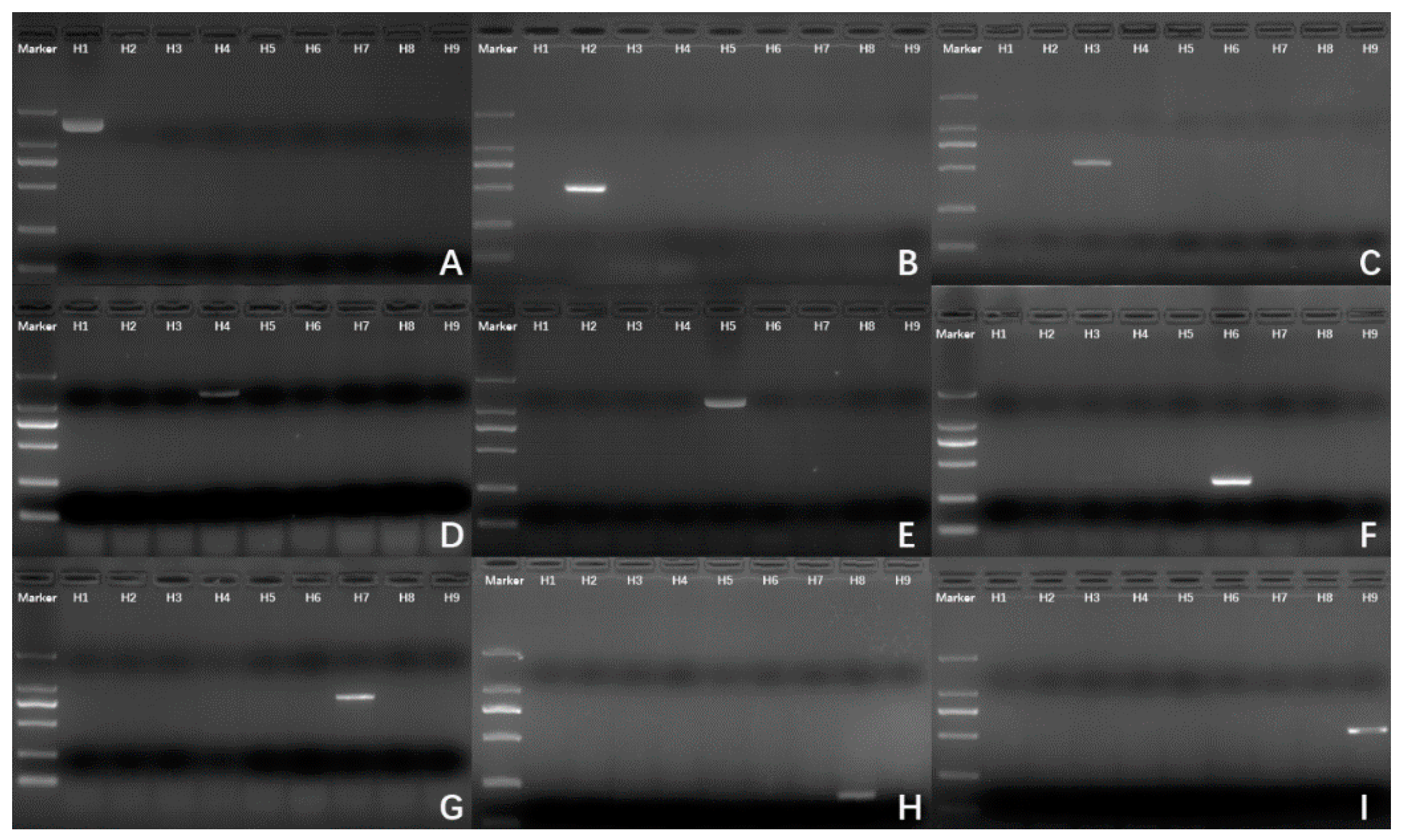
3.2. PCR Experiment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Short, K.R.; Kedzierska, K.; van de Sandt, C.E. Back to the Future: Lessons Learned from the 1918 Influenza Pandemic. Front. Cell. Infect. Microbiol. 2018, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Ask the Expert: Influenza Q&A. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 6 November 2018).
- Chiu, W.; Burnett, R.; Garcea, R. Attachment and entry of influenza virus into host cells. Pivotal roles of hemagglutinin. In Structural Biology of Viruses; Chiu, W., Burnett, R.M., Garcea, R.L., Eds.; Oxford University Press: New York, NY, USA, 1997; pp. 80–104. [Google Scholar]
- Ketklao, S.; Ketklao, S.; Boonarkart, C.; Phakaratsakul, S.; Auewarakul, P.; Suptawiwat, O. Responses to the Sb epitope contributed to antigenic drift of the influenza A 2009 H1N1 virus. Arch. Virol. 2020, 165, 2503–2512. [Google Scholar] [CrossRef]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- Mayr, J.; Lau, K.; Lai, J.C.C.; Gagarinov, I.A.; Shi, Y.; McAtamney, S.; Chan, R.W.Y.; Nicholls, J.; von Itzstein, M.; Haselhorst, T. Unravelling the Role of O-glycans in Influenza A Virus Infection. Sci. Rep. 2018, 8, 16382. [Google Scholar] [CrossRef]
- Khanna, M.; Sharma, S.; Kumar, B.; Rajput, R. Protective immunity based on the conserved hemagglutinin stalk domain and its prospects for universal influenza vaccine development. BioMed Res. Int. 2014, 2014, 546274. [Google Scholar] [CrossRef] [PubMed]
- Giles, B.M.; Ross, T.M. A computationally optimized broadly reactive antigen (COBRA) based H5N1 VLP vaccine elicits broadly reactive antibodies in mice and ferrets. Vaccine 2011, 29, 3043–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, R.G.; Govorkova, E.A. Continuing Challenges in Influenza. Ann. N. Y. Acad. Sci. 2014, 1323, 115–139. [Google Scholar] [CrossRef]
- Sisteré-Oró, M.; Lopez-Serrano, S.; Veljkovic, V.; Pina-Pedrero, S.; Vergara-Alert, J.; Cordoba, L.; Perez-Maillo, M.; Pleguezuelos, P.; Vidal, E.; Segales, J.; et al. DNA vaccine based on conserved HA-peptides induces strong immune response and rapidly clears influenza virus infection from vaccinated pigs. PLoS ONE 2019, 14, e0222201. [Google Scholar] [CrossRef]
- Estrada, L.D.; Schultz-Cherry, S. Development of a Universal Influenza Vaccine. J. Immunol. 2019, 202, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to search for similarities in the amino acid sequence of 2 proteins. J. Mol. Biol. 1970, 48, 443. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.-M.; Taly, J.-F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 2008, 82, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Zeng, Y. Comparative study of different nucleic acid extraction methods for influenza a H1N1 virus. J. Clin. Exp. Med. 2017, 16, 230–232. [Google Scholar]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The Structure and Receptor Binding Properties of the 1918 Influenza Hemagglutinin. Science 2004, 303, 1838. [Google Scholar] [CrossRef]
- Liu, J.; Stevens, D.J.; Haire, L.F.; Walker, P.A.; Coombs, P.J.; Russell, R.J.; Gamblin, S.J.; Skehel, J.J. Structures of receptor complexes formed by hemagglutinins from the Asian Influenza pandemic of 1957. Proc. Natl. Acad. Sci. USA 2009, 106, 17175–17180. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.J.; Vachieri, S.G.; Haire, L.F.; Ogrodowicz, R.W.; Martin, S.R.; Walker, P.A.; Xiong, X.; Gamblin, S.J.; Skehel, J.J. Recent evolution of equine influenza and the origin of canine influenza. Proc. Natl. Acad. Sci. USA 2014, 111, 11175–11180. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Qi, J.; Xiao, H.; Bi, Y.; Zhang, W.; Xu, Y.; Wang, F.; Shi, Y.; Gao, G.F. Avian-to-Human Receptor-Binding Adaptation by Influenza A Virus Hemagglutinin H4. Cell Rep. 2017, 20, 1201–1214. [Google Scholar] [CrossRef] [Green Version]
- Ha, Y.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. H5 avian and H9 swine influenza virus haemagglutinin structures: Possible origin of influenza subtypes. EMBO J. 2002, 21, 865–875. [Google Scholar] [CrossRef] [Green Version]
- de Vries, R.P.; Tzarum, N.; Peng, W.; Thompson, A.J.; Ambepitiya Wickramasinghe, I.N.; de la Pena, A.T.T.; van Breemen, M.J.; Bouwman, K.M.; Zhu, X.; McBride, R.; et al. A single mutation in Taiwanese H6N1 influenza hemagglutinin switches binding to human-type receptors. EMBO Mol. Med. 2017, 9, 1314–1325. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.J.; Gamblin, S.J.; Haire, L.F.; Stevens, D.J.; Xiao, B.; Ha, Y.; Skehel, J.J. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology 2004, 325, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitschi, F.; Devauchelle, C.; Corel, E. Automatic detection of anchor points for multiple sequence alignment. BMC Bioinform. 2010, 11, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duh, D.; Blažič, B. Single mutation in the matrix gene of seasonal influenza A viruses critically affects the performance of diagnostic molecular assay. J. Virol. Methods 2018, 251, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. Novel universal influenza virus vaccine approaches. Curr. Opin. Virol. 2016, 17, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Freyn, A.W.; da Silva, J.R.; Rosado, V.C.; Bliss, C.M.; Pine, M.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; de Souza Ferreira, L.C.; Weissman, D.; et al. A Multi-Targeting, Nucleoside-Modified mRNA Influenza Virus Vaccine Provides Broad Protection in Mice. Mol. Ther. 2020, 28, 1569–1584. [Google Scholar] [CrossRef]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Kariko, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 2018, 9, 3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Rodriguez, J.; Meijerhof, T.; Niesters, H.G.; Stjernholm, G.; Hovden, A.-O.; Sorensen, B.; Okvist, M.; Sommerfelt, M.A.; Huckriede, A. A novel peptide-based vaccine candidate with protective efficacy against influenza A in a mouse model. Virology 2018, 515, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.V.; Khrustaleva, T.A.; Kordyukova, L.V. Selection and structural analysis of the NY25 peptide—A vaccine candidate from hemagglutinin of swine-origin Influenza H1N1. Microb. Pathog. 2018, 125, 72–83. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | Nucleotide | Protein |
|---|---|---|
| H1 | 23,543 | 19,916 |
| H2 | 613 | 624 |
| H3 | 19,358 | 21,743 |
| H4 | 1868 | 1896 |
| H5 | 5658 | 6325 |
| H6 | 1745 | 1788 |
| H7 | 2090 | 2203 |
| H8 | 139 | 141 |
| H9 | 3623 | 3718 |
| Subtype | Description | Catalog Number |
|---|---|---|
| H1 | H1N1 (A/Beijing/262/1995) Hemagglutinin | VG11068-UT |
| H2 | H2N2 (A/Guiyang/1/1957) Hemagglutinin | VG40119-UT |
| H3 | H3N2 (A/Hong Kong/1/1968) Hemagglutinin | VG40116-UT |
| H4 | H4N6 (A/Swine/Ontario/01911-1/99) Hemagglutinin | VG11706-UT |
| H5 | H5N1 (Anhui/1/2005) Hemagglutinin | VG11048-UT |
| H6 | H6N2 (A/chicken/Guangdong/C273/2011) Hemagglutinin | VG40398-UT |
| H7 | H7N9 (A/Hangzhou/1/2013) Hemagglutinin | VG40105-UT |
| H8 | H8N4 (A/pintail duck/Alberta/114/1979) Hemagglutinin | VG11722-UT |
| H9 | H9N2 (A/Chicken/Hong Kong/G9/97) Hemagglutinin | VG40036-UT |
| Subtype | Protein Sequence | Primer Sequence | Fragment Length(bp) |
|---|---|---|---|
| H1 | NVTVTHS | (5′–3′)AATGTRACWGTRACMCACTCW | 1534 |
| SFWMCSN | (3′–5′)ATTRGARCACATCCARAARCT | ||
| H2 | YHHSNDQ | (5′–3′)TAYCAYCACAGCAATGAYCAR | 481 |
| YQILAIYAT | (3′–5′)TGTAGCRTADATDGCAAGDATTTGRTA | ||
| H3 | ITPNGSI | (5′–3′)ATYACTCCAAATGGAAGCATY | 532 |
| AEDMGN | (3′–5′)ATTKCCCATRTCYTCAGC | ||
| H4 | CYPFDV | (5′–3′)TGYTAYCCATTTGATGTG | 1243 |
| QGYKDI | (3′–5′)RATGTCYTTGTATCCYTG | ||
| H5 | VTVTHA | (5′–3′)GTBACKGTYACACAYGCY | 1219 |
| LMENERTLD | (3′–5′)RTCYAGAGTTCTYTCATTTTCCATGAG | ||
| H6 | WYGYHHE | (5′–3′)TGGTAYGGMTAYCAYCATGAR | 349 |
| CFEFWHKC | (3′–5′)RCAYTTRTGCCARAATTCAAARCA | ||
| H7 | FYAEMK | (5′–3′)TTCTATGCRGARATGAAR | 790 |
| GNVINW | (3′–5′)CCARTTWATSACATTVCC | ||
| H8 | EGMCYP | (5′–3′)GAGGGRATGTGYTAYCCT | 175 |
| SINWLTKK | (3′–5′)CTTYTTRGTYARCCARTTRATGCT | ||
| H9 | GWYGFQHS | (5′–3′)GGTTGGTATGGDTTCCAGCATTCA | 556 |
| AFLFWAM | (3′–5′)CATGGCCCAGAAYARGAAGGC |
| Subtype | Step 1 | Step 2 | Step 3 | Cycles | ||
|---|---|---|---|---|---|---|
| S1 | S2 | S3 | ||||
| H1 | 94 °C 5 min | 94 °C 30 s | 56 °C/2 min | 72 °C/90 s | 72 °C 7 min | 30 |
| H2 | 58.5 °C/1 min | 72 °C/70 s | ||||
| H3 | 59 °C/1 min | 72 °C/30 s | ||||
| H4 | 58.5 °C/2 min | 72 °C/70 s | ||||
| H5 | 59 °C/2 min | 72 °C/70 s | ||||
| H6 | 58 °C/30 s | 72 °C/30 s | ||||
| H7 | 50 °C/1 min | 72 °C/30 s | ||||
| H8 | 59 °C/30 s | 72 °C/70 s | ||||
| H9 | 65 °C/1 min | 72 °C/30 s | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Q.; Ji, X.; Wu, F.; Ma, L. Conserved Sequence Analysis of Influenza A Virus HA Segment and Its Application in Rapid Typing. Diagnostics 2021, 11, 1328. https://doi.org/10.3390/diagnostics11081328
Lin Q, Ji X, Wu F, Ma L. Conserved Sequence Analysis of Influenza A Virus HA Segment and Its Application in Rapid Typing. Diagnostics. 2021; 11(8):1328. https://doi.org/10.3390/diagnostics11081328
Chicago/Turabian StyleLin, Qianyu, Xiang Ji, Feng Wu, and Lan Ma. 2021. "Conserved Sequence Analysis of Influenza A Virus HA Segment and Its Application in Rapid Typing" Diagnostics 11, no. 8: 1328. https://doi.org/10.3390/diagnostics11081328
APA StyleLin, Q., Ji, X., Wu, F., & Ma, L. (2021). Conserved Sequence Analysis of Influenza A Virus HA Segment and Its Application in Rapid Typing. Diagnostics, 11(8), 1328. https://doi.org/10.3390/diagnostics11081328