Sudden Cardiac Death Caused by a Fatal Association of Hypertrophic Cardiomyopathy (MYH7, p.Arg719Trp), Heterozygous Familial Hypercholesterolemia (LDLR, p.Gly343Lys) and SARS-CoV-2 B.1.1.7 Infection

 ,
,  , ,
, ,
Abstract
:1. Introduction
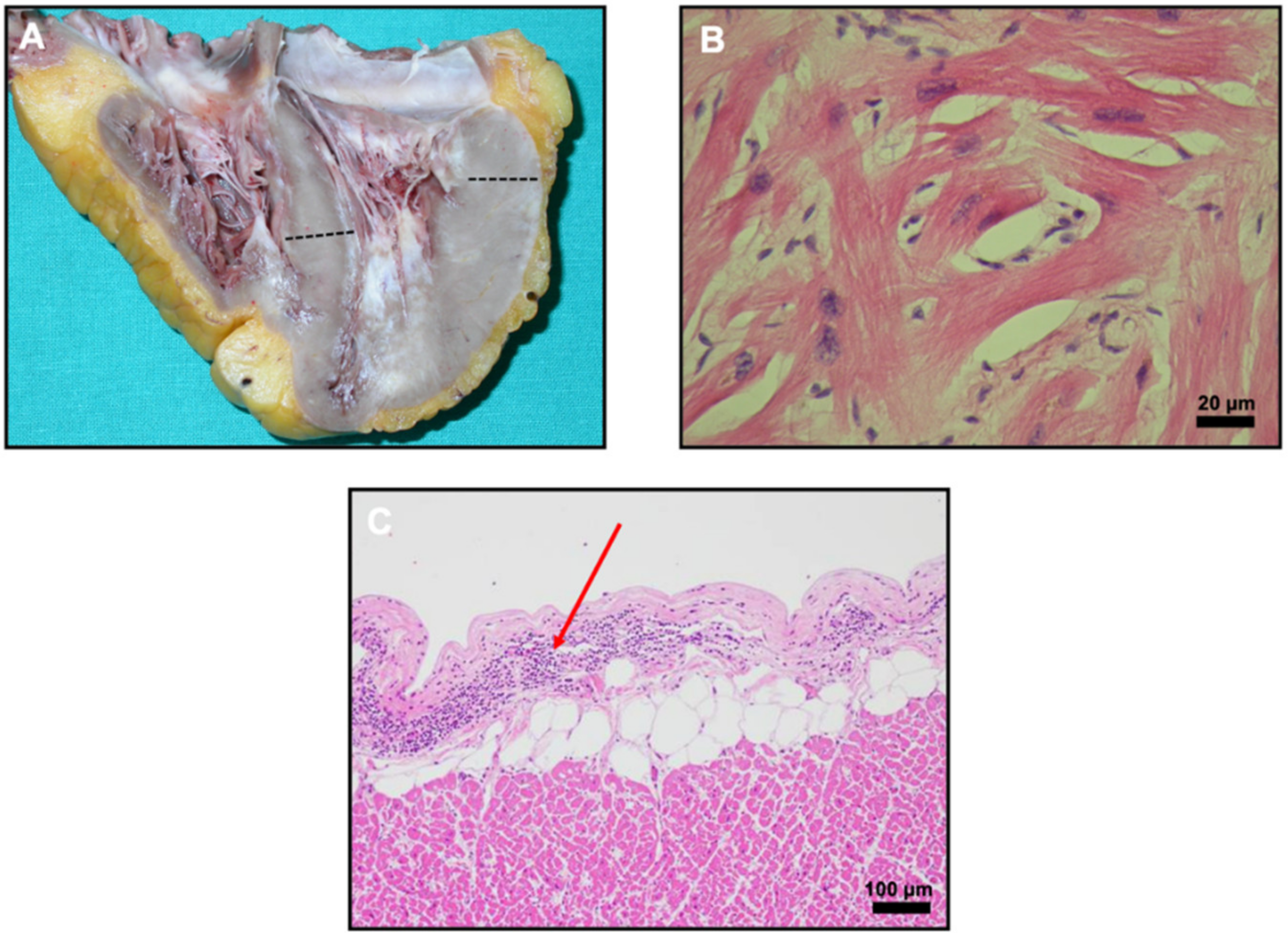
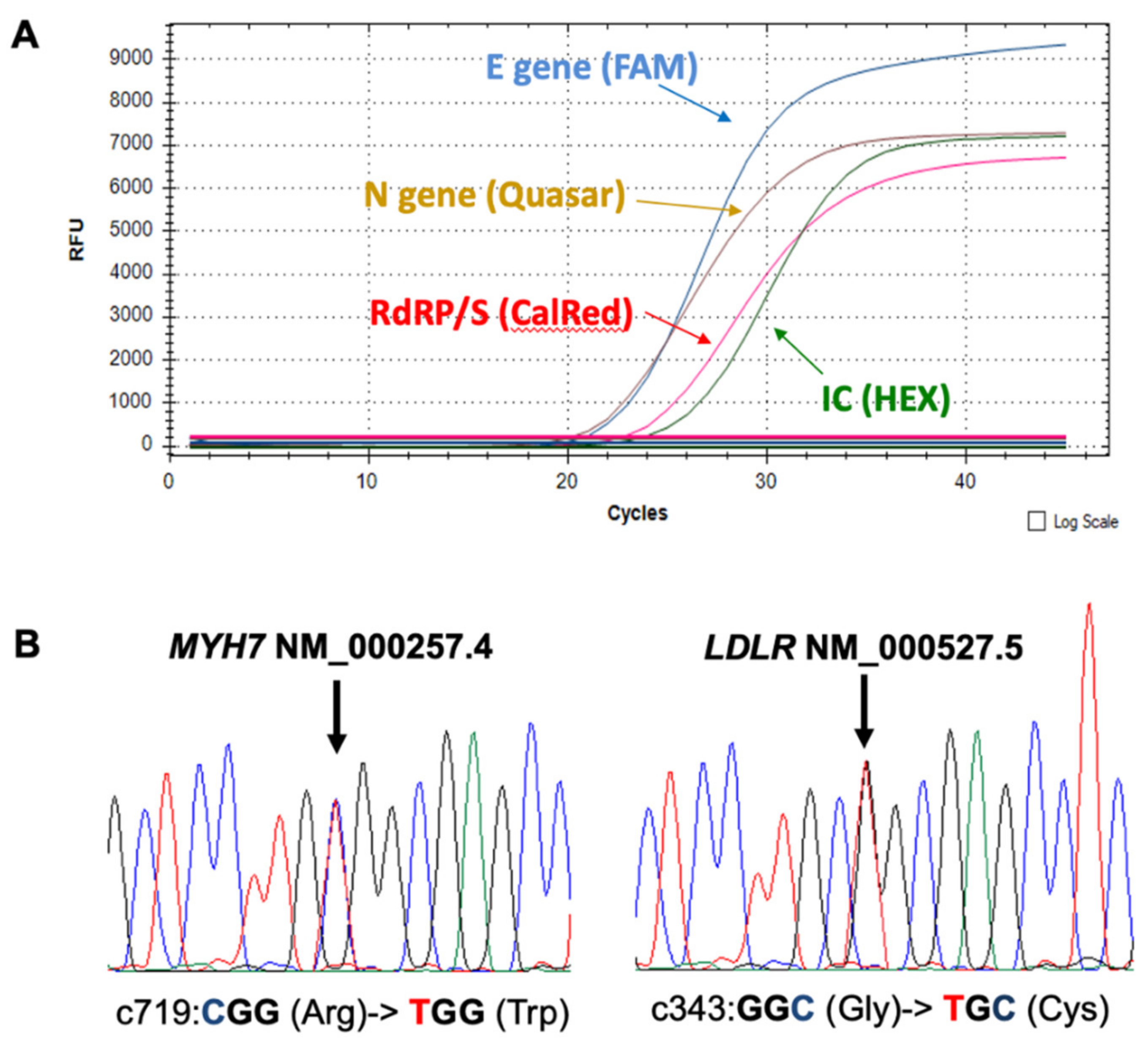
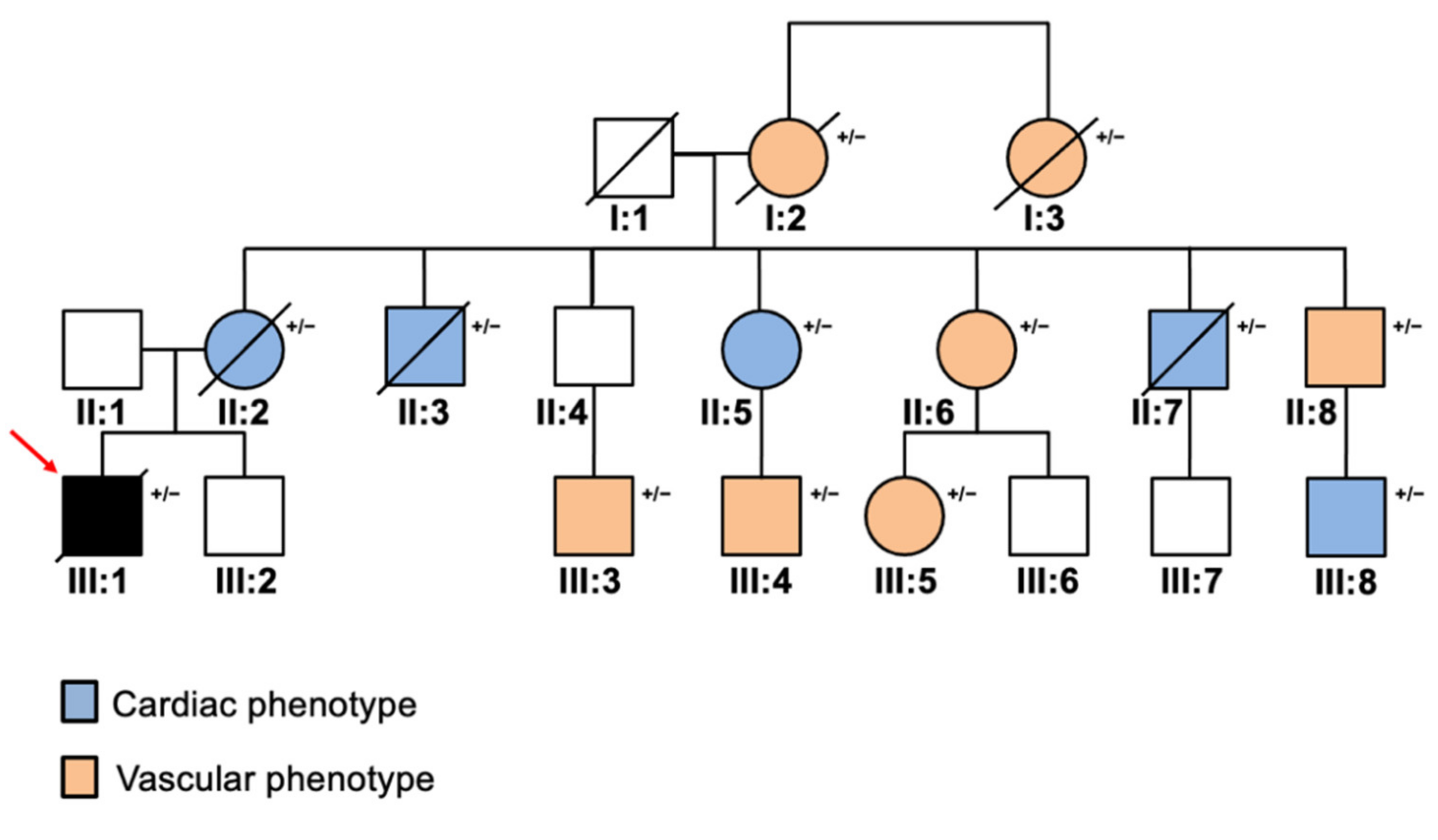
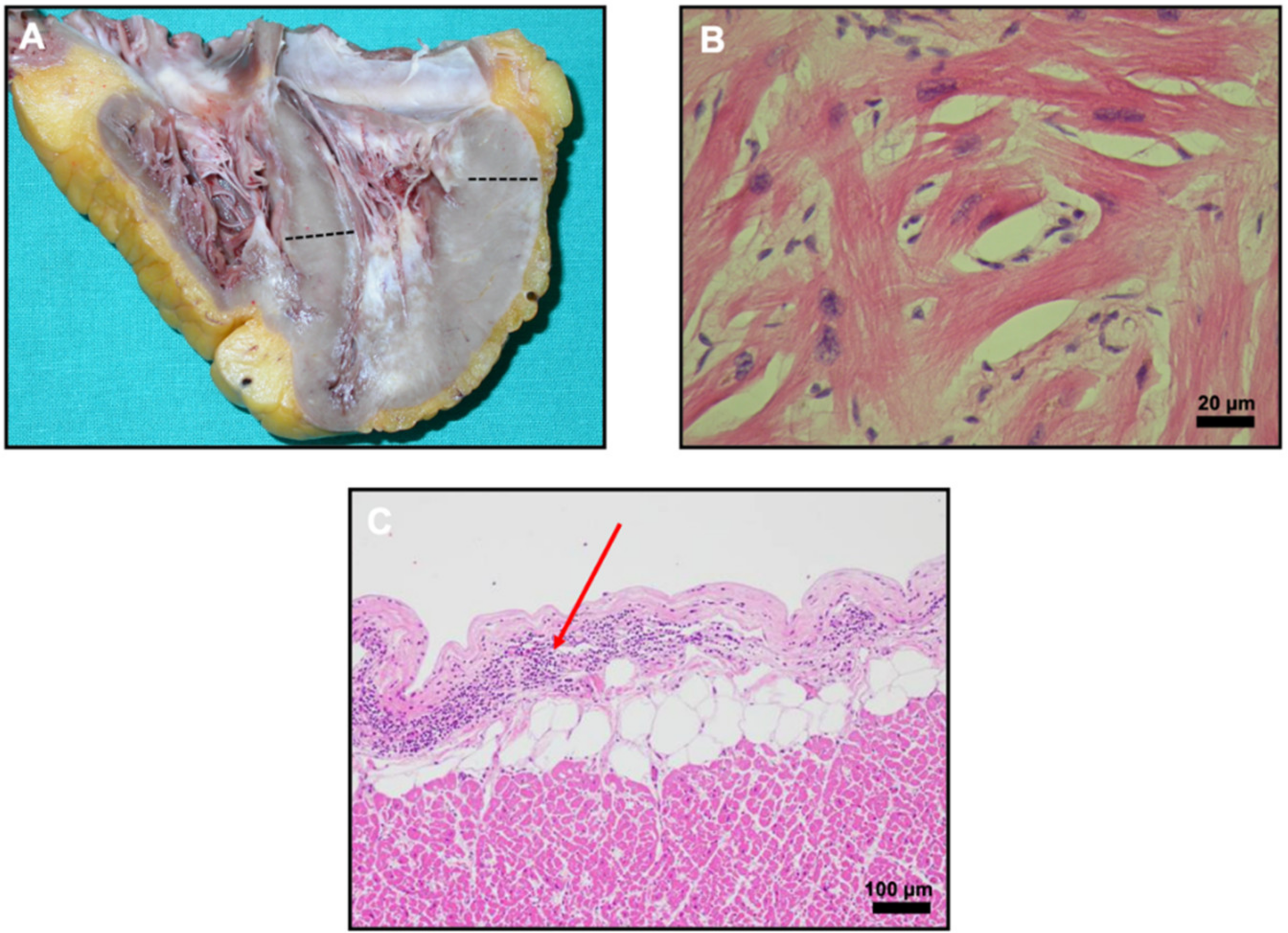
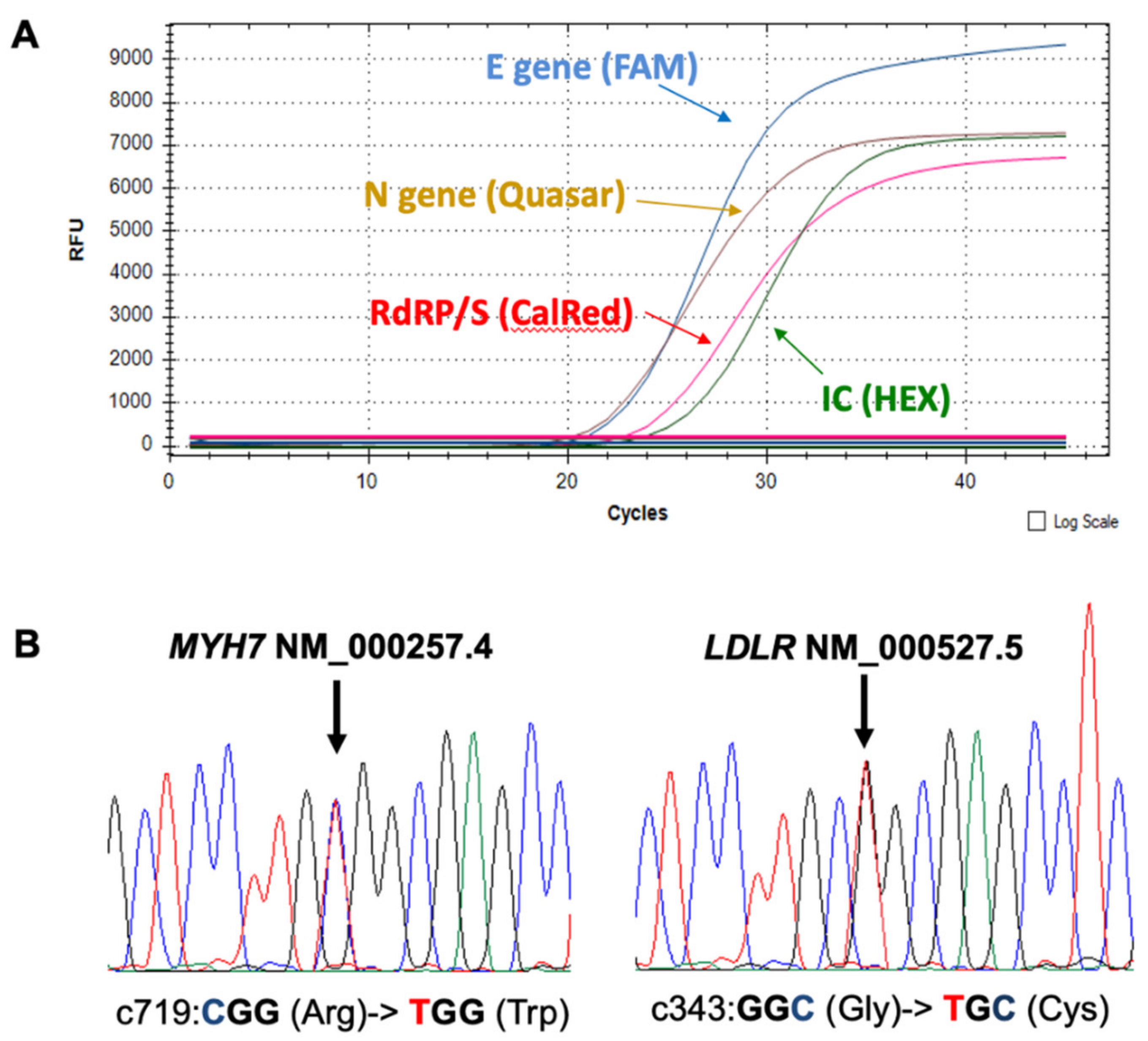
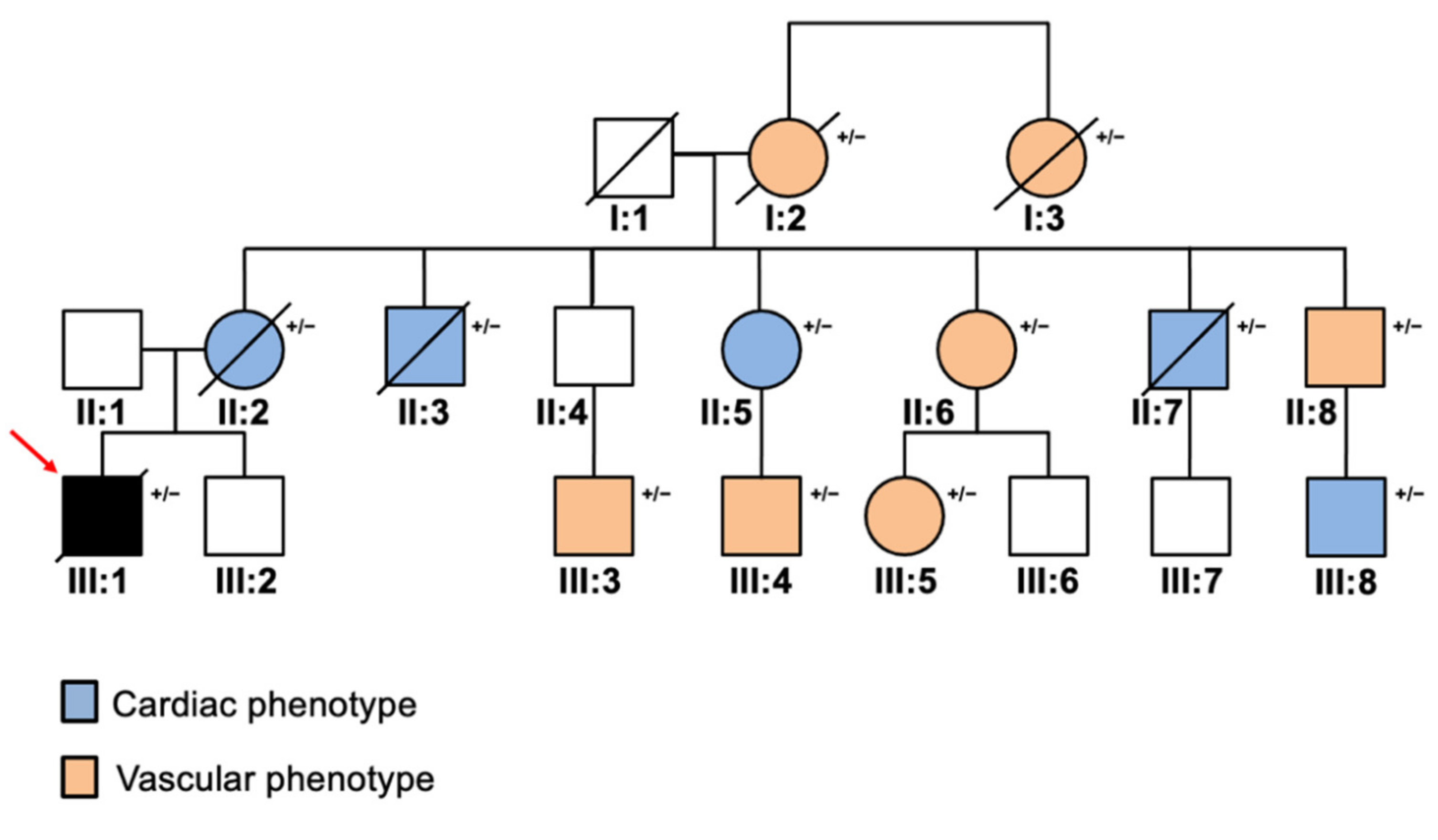
2. Case Report
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; Kondapally Seshasai, S.R.; Cole, D.; Hovingh, G.K.; Kastelein, J.J.P.; Mata, P.; Raal, F.J.; Santos, R.D.; Soran, H.; Watts, G.F.; et al. Familial hypercholesterolaemia: A global call to arms. Atherosclerosis 2015, 243, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- McGowan, M.P.; Hosseini Dehkordi, S.H.; Moriarty, P.M.; Duell, P.B. Diagnosis and Treatment of Heterozygous Familial Hypercholesterolemia. J. Am. Heart Assoc. 2019, 8, e013225. [Google Scholar] [CrossRef]
- Basso, C.; Leone, O.; Rizzo, S.; De Gaspari, M.; Van Der Wal, A.C.; Aubry, M.C.; Bois, M.C.; Lin, P.T.; Maleszewski, J.J.; Stone, J.R. Pathological features of COVID-19-associated myocardial injury: A multicentre cardiovascular pathology study. Eur. Heart J. 2020, 41, 3827–3835. [Google Scholar] [CrossRef]
- Bearse, M.; Hung, Y.P.; Krauson, A.J.; Bonanno, L.; Boyraz, B.; Harris, C.K.; Helland, T.L.; Hilburn, C.F.; Hutchison, B.; Jobbagy, S.; et al. Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod. Pathol. 2021, 34, 1345–1357. [Google Scholar] [CrossRef]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1084–H1090. [Google Scholar] [CrossRef] [Green Version]
- Pirzada, A.; Mokhtar, A.T.; Moeller, A.D. COVID-19 and Myocarditis: What Do We Know So Far? CJC Open 2020, 2, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Egbuche, O.; Igwe, J.; Jegede, O.; Wagle, B.; Olanipekun, T.; Onwuanyi, A. Cardiovascular complications in COVID-19 patients with or without diabetes mellitus. Endocrinol. Diabetes Metab. 2021, 4, e00218. [Google Scholar] [CrossRef] [PubMed]
- Shchendrygina, A.; Nagel, E.; Puntmann, V.O.; Valbuena-Lopez, S. COVID-19 myocarditis and prospective heart failure burden. Expert Rev. Cardiovasc. Ther. 2021, 19, 5–14. [Google Scholar] [CrossRef]
- Bansal, M. Cardiovascular disease and COVID-19. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.Y.; Redwood, S.; Prendergast, B.; Chen, M. Coronaviruses and the cardiovascular system: Acute and long-term implications. Eur. Heart J. 2020, 41, 1798–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Anan, R.; Greve, G.; Thierfelder, L.; Watkins, H.; McKenna, W.J.; Solomon, S.; Vecchio, C.; Shono, H.; Nakao, S.; Tanaka, H.; et al. Prognostic implications of novel β cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J. Clin. Investig. 1994, 93, 280–285. [Google Scholar] [CrossRef]
- Jelassi, A.; Jguirim, I.; Najah, M.; Abid, A.M.; Boughamoura, L.; Maatouk, F.; Rouis, M.; Boileau, C.; Rabès, J.P.; Slimane, M.N.; et al. Limited mutational heterogeneity in the LDLR gene in familial hypercholesterolemia in Tunisia. Atherosclerosis 2009, 203, 449–453. [Google Scholar] [CrossRef]
- Lara, D.; Young, T.; Del Toro, K.; Chan, V.; Ianiro, C.; Hunt, K.; Kleinmahon, J. Acute fulminant myocarditis in a pediatric patient with covid-19 infection. Pediatrics 2020, 146, e20201509. [Google Scholar] [CrossRef] [PubMed]
- Gauchotte, G.; Venard, V.; Segondy, M.; Cadoz, C.; Esposito-Fava, A.; Barraud, D.; Louis, G. SARS-Cov-2 fulminant myocarditis: An autopsy and histopathological case study. Int. J. Legal Med. 2021, 135, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Torres, W.; Herrera-Escandón, Á.; Hurtado-Rivera, M.; Plata-Mosquera, C.A. COVID-19 fulminant myocarditis: A case report. Eur. Heart J. Case Rep. 2020, 4, 1–6. [Google Scholar] [CrossRef]
- Zeng, J.H.; Liu, Y.X.; Yuan, J.; Wang, F.X.; Wu, W.B.; Li, J.X.; Wang, L.F.; Gao, H.; Wang, Y.; Dong, C.F.; et al. First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection 2020, 48, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Kesici, S.; Aykan, H.H.; Orhan, D.; Bayrakci, B. Fulminant COVID-19-related myocarditis in an infant. Eur. Heart J. 2020, 41, 3021. [Google Scholar] [CrossRef]
- Naneishvili, T.; Khalil, A.; O’Leary, R.; Prasad, N. Fulminant myocarditis as an early presentation of SARS-CoV-2. BMJ Case Rep. 2020, 13, 237553. [Google Scholar] [CrossRef]
- Garot, J.; Amour, J.; Pezel, T.; Dermoch, F.; Messadaa, K.; Felten, M.-L.; Raymond, V.; Baubillier, E.; Sanguineti, F.; Garot, P. SARS-CoV-2 Fulminant Myocarditis. JACC Case Rep. 2020, 2, 1342–1346. [Google Scholar] [CrossRef]
- Wenzel, P.; Kopp, S.; Gobel, S.; Jansen, T.; Geyer, M.; Hahn, F.; Kreitner, K.F.; Escher, F.; Schultheiss, H.P.; Münzel, T. Evidence of SARS-CoV-2 mRNA in endomyocardial biopsies of patients with clinically suspected myocarditis tested negative for COVID-19 in nasopharyngeal swab. Cardiovasc. Res. 2020, 116, 1661–1663. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Schaller, T.; Hirschbühl, K.; Burkhardt, K.; Braun, G.; Trepel, M.; Märkl, B.; Claus, R. Postmortem Examination of Patients with COVID-19. JAMA 2020, 323, 2518. [Google Scholar] [CrossRef]
- Tavazzi, G.; Pellegrini, C.; Maurelli, M.; Belliato, M.; Sciutti, F.; Bottazzi, A.; Sepe, P.A.; Resasco, T.; Camporotondo, R.; Bruno, R.; et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 2020, 22, 911–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential Effects of Coronaviruses on the Cardiovascular System: A Review. JAMA Cardiol. 2020, 5, 831–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiq, M.M.; Chan, A.T.; Miorin, L.; Yadaw, A.S.; Beaumont, K.G.; Kehrer, T.; White, K.M.; Cupic, A.; Tolentino, R.E.; Hu, B.; et al. Physiology of cardiomyocyte injury in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, P.; Khanji, M.Y.; Cooper, L.T.; Chahal, C.A.A. Recognizing COVID-19–related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 2020, 17, 1463–1471. [Google Scholar] [CrossRef]
- Petrovic, V.; Radenkovic, D.; Radenkovic, G.; Djordjevic, V.; Banach, M. Pathophysiology of Cardiovascular Complications in COVID-19. Front. Physiol. 2020, 11, 575600. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, K.A.; Meyer-Schwickerath, C.; Heger, E.; Knops, E.; Lehmann, C.; Rybniker, J.; Schommers, P.; Eichenauer, D.A.; Kurth, F.; Ramharter, M.; et al. RNAemia Corresponds to Disease Severity and Antibody Response in Hospitalized COVID-19 Patients. Viruses 2020, 12, 1045. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Desai, R.; Gandhi, Z.; Fong, H.K.; Doreswamy, S.; Desai, V.; Chockalingam, A.; Mehta, P.K.; Sachdeva, R.; Kumar, G. Takotsubo Syndrome in Patients with COVID-19: A Systematic Review of Published Cases. SN Compr. Clin. Med. 2020, 2, 2102–2108. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Elliott, P.; McKenna, W. Sudden cardiac death in hypertrophic cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2013, 6, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuorio, A.; Watts, G.F.; Kovanen, P.T. Familial hypercholesterolaemia and COVID-19: Triggering of increased sustained cardiovascular risk. J. Intern. Med. 2020, 287, 746–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, O.; Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Cesar, S.; Mademont, I.; Mates, J.; Pérez-Serra, A.; Coll, M.; Pico, F.; et al. Natural and undetermined sudden death: Value of post-mortem genetic investigation. PLoS ONE 2016, 11, e0167358. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Genes Included in the Custom Cardio-Panel |
|---|
| ACTC1, ACVRL1, APOB, BAG3, BMPR2, BRAF, CACNA1C, CASQ2, DES, DMD, DSC2, DSG2, DSP, ELN, EMD, ENG, FBN1, FLNC, GATA4, GLA, JAG1, JUP, KCNE1, KCNE2, KCNH2, KCNJ2, KCNJ8, KCNQ1, KRAS, LAMP2, LDLR, LDLRAP1, LMNA, MYBPC3, MYH7, MYL2, MYL3, NF1, NKX2-5, PKP2, PLN, PRKAG2, PCSK9, PTPN11, RAF1, RBM20, RYR2, SCN1B, SCN5A, SOS1, SOS2, TAZ, TGFBR2, TMEM43, TNNC1, TNNI3, TNNT2, TPM1, TTN, TTR |
| ID | Age | Sex | Genotype | sLDL Levels (mg/dL) * | Phenotype | |
|---|---|---|---|---|---|---|
| Cardiac | Vascular | |||||
| I:2 | 50 † | F | LDLR +/− | 182 | CAD | - |
| I:3 | 55 † | F | N.A. | 210 | CAD | - |
| II:2 | 60 † | F | LDLR +/− | 197 | - | ECO |
| II:3 | 58 † | M | LDLR +/− | 221 | - | ECO |
| II:5 | 56 | F | LDLR +/− | 230 | - | ECO |
| II:6 | 54 | F | LDLR +/− | 227 | CAD | - |
| II:7 | 50 † | M | LDLR +/− | 248 | - | ECO |
| II:8 | 48 | M | LDLR +/− | 201 | CAD | - |
| III:1 | 32 † | M | LDLR +/−; MYH7 +/− | 248 | CAD, LVH | - |
| III:3 | 28 | M | LDLR +/− | 278 | CAD | - |
| III:4 | 27 | M | LDLR +/− | 297 | CAD | - |
| III:5 | 27 | F | LDLR +/− | 289 | CAD | - |
| III:8 | 28 | M | LDLR +/− | 211 | - | ECO |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marziliano, N.; Medoro, A.; Mignogna, D.; Saccon, G.; Folzani, S.; Reverberi, C.; Russo, C.; Intrieri, M. Sudden Cardiac Death Caused by a Fatal Association of Hypertrophic Cardiomyopathy (MYH7, p.Arg719Trp), Heterozygous Familial Hypercholesterolemia (LDLR, p.Gly343Lys) and SARS-CoV-2 B.1.1.7 Infection. Diagnostics 2021, 11, 1229. https://doi.org/10.3390/diagnostics11071229
Marziliano N, Medoro A, Mignogna D, Saccon G, Folzani S, Reverberi C, Russo C, Intrieri M. Sudden Cardiac Death Caused by a Fatal Association of Hypertrophic Cardiomyopathy (MYH7, p.Arg719Trp), Heterozygous Familial Hypercholesterolemia (LDLR, p.Gly343Lys) and SARS-CoV-2 B.1.1.7 Infection. Diagnostics. 2021; 11(7):1229. https://doi.org/10.3390/diagnostics11071229
Chicago/Turabian StyleMarziliano, Nicola, Alessandro Medoro, Donatella Mignogna, Giovanni Saccon, Stefano Folzani, Claudio Reverberi, Claudio Russo, and Mariano Intrieri. 2021. "Sudden Cardiac Death Caused by a Fatal Association of Hypertrophic Cardiomyopathy (MYH7, p.Arg719Trp), Heterozygous Familial Hypercholesterolemia (LDLR, p.Gly343Lys) and SARS-CoV-2 B.1.1.7 Infection" Diagnostics 11, no. 7: 1229. https://doi.org/10.3390/diagnostics11071229
APA StyleMarziliano, N., Medoro, A., Mignogna, D., Saccon, G., Folzani, S., Reverberi, C., Russo, C., & Intrieri, M. (2021). Sudden Cardiac Death Caused by a Fatal Association of Hypertrophic Cardiomyopathy (MYH7, p.Arg719Trp), Heterozygous Familial Hypercholesterolemia (LDLR, p.Gly343Lys) and SARS-CoV-2 B.1.1.7 Infection. Diagnostics, 11(7), 1229. https://doi.org/10.3390/diagnostics11071229