
Implementation of Precision Oncology for Patients with Metastatic Breast Cancer in an Interdisciplinary MTB Setting

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Study Design
2.2. Panel-Guided Next-Generation Sequencing
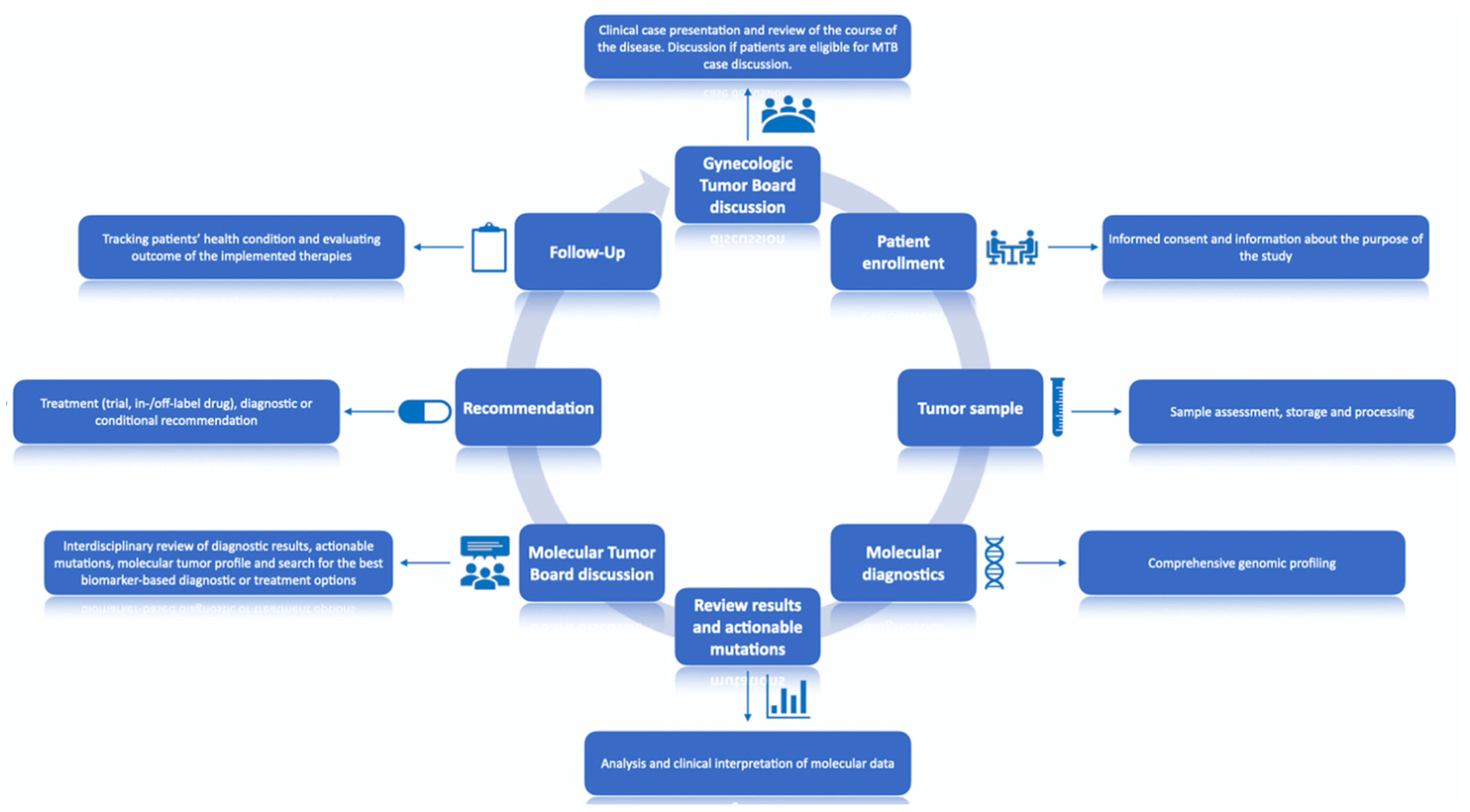
2.3. Study Procedure
2.4. Analysis of Results
3. Results
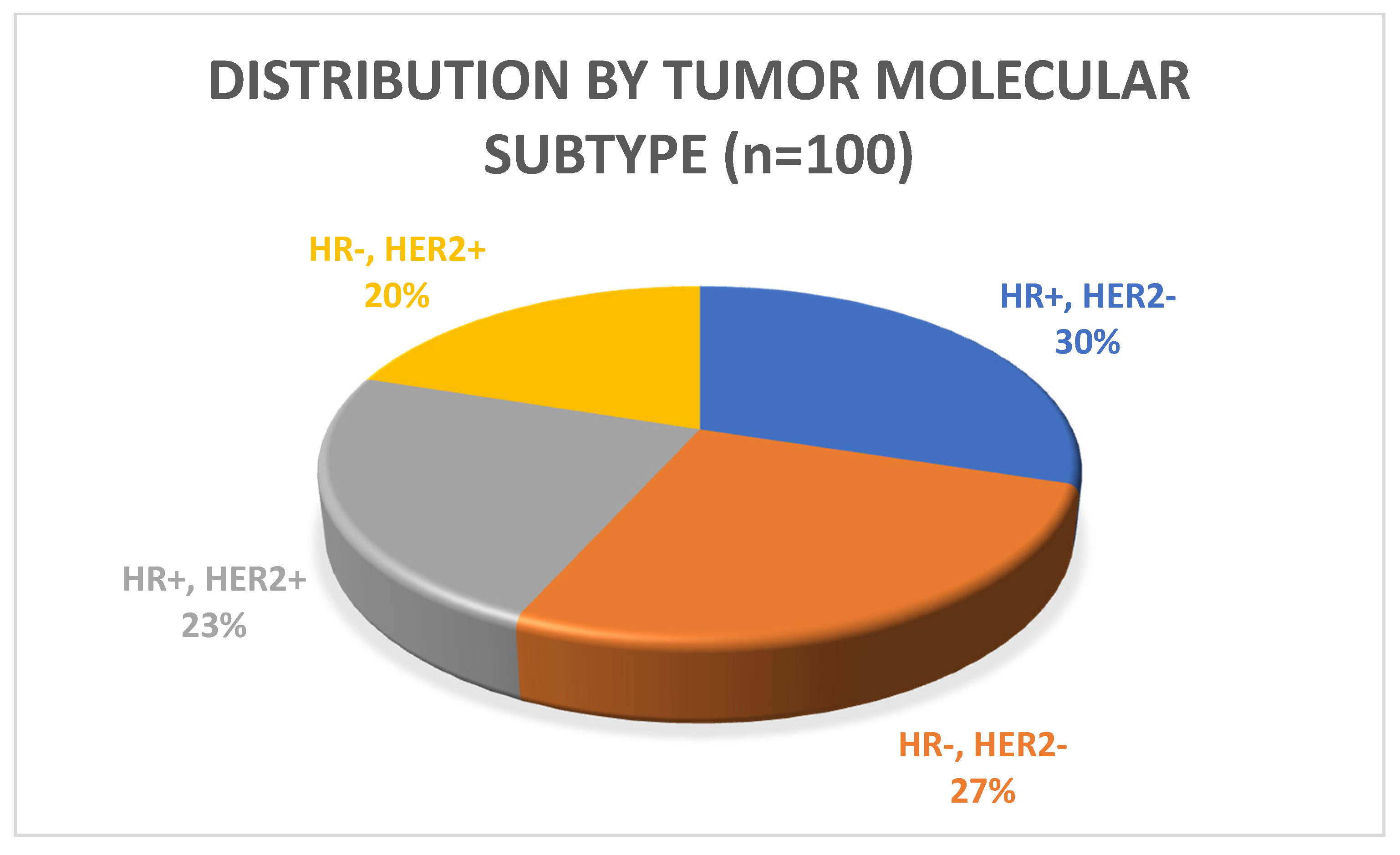
3.1. Patient Characteristics
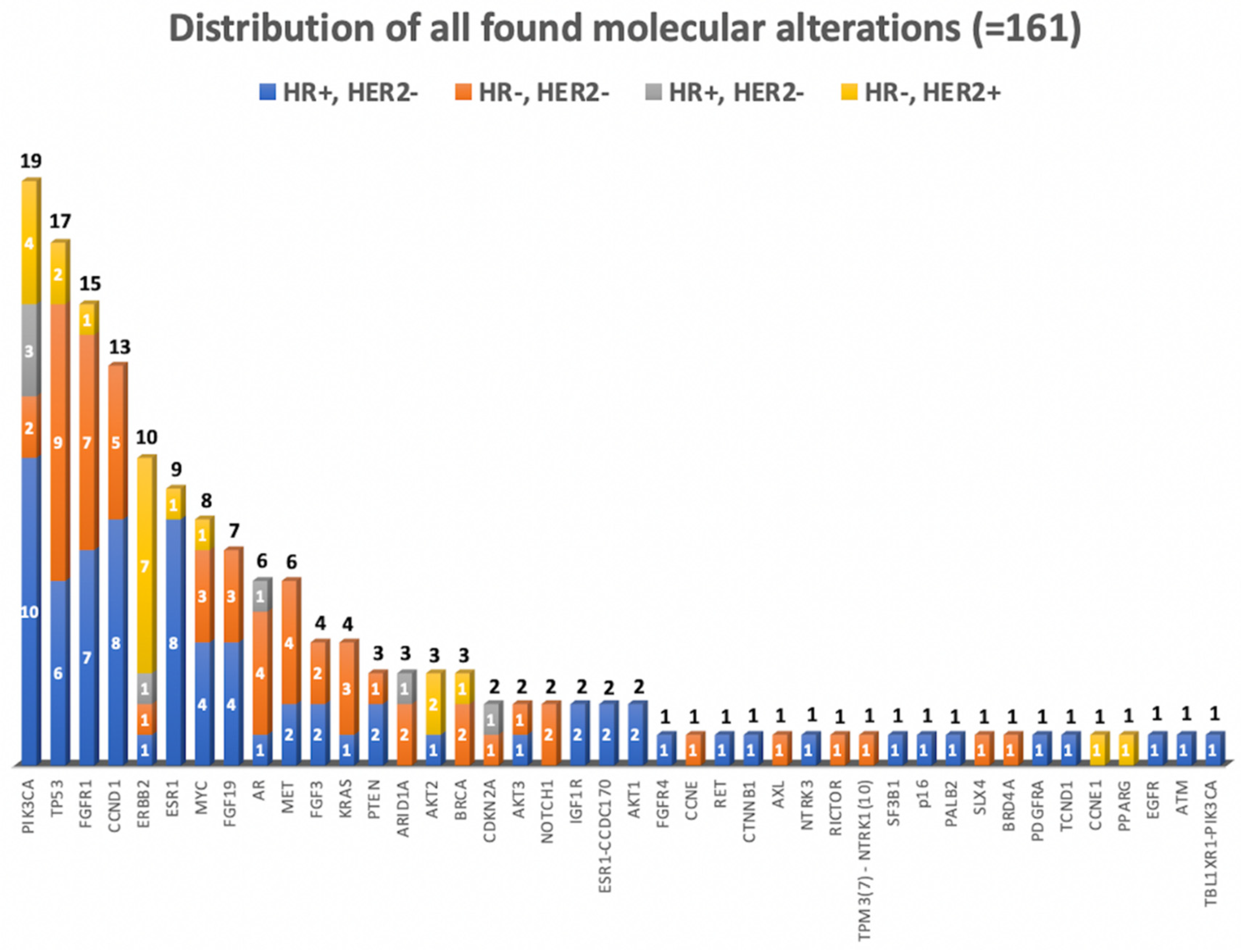
3.2. Molecular Diagnostics
3.3. Recommendations
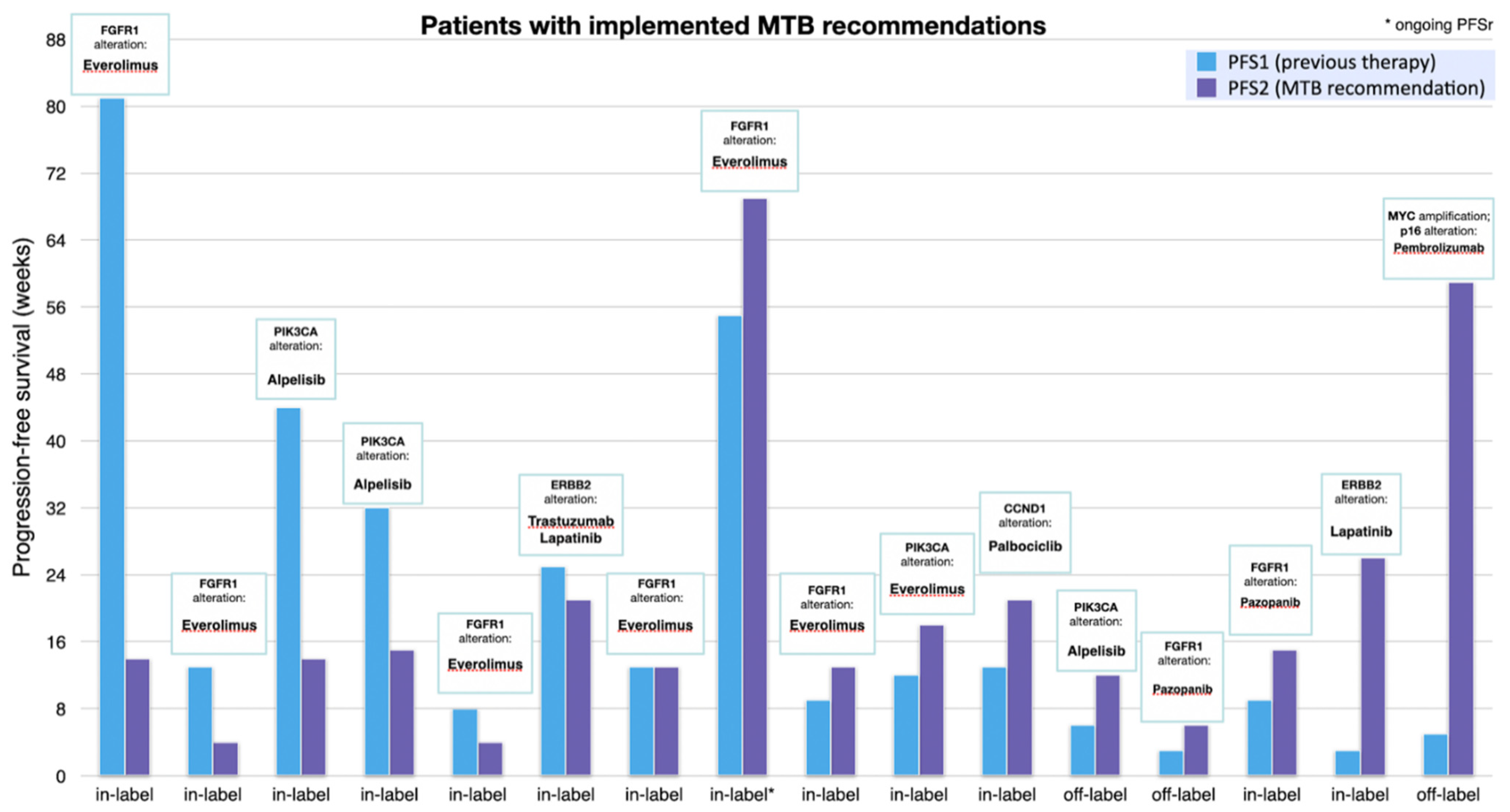
3.4. Progression Free Survival Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Gene Alteration | Implemented Therapy (MTB Recommendation) | Previous Therapy | Label | PFS2 (Weeks) | PFS1 (Weeks) | PFSr |
| 1 | FGFR1 | Everolimus [31,32] | Capecitabine | in | 14 | 81 | 0.2 |
| 2 | FGFR1 | Everolimus | Capecitabine/Bevacizumab | in | 4 | 13 | 0.3 |
| 3 | PIK3CA | Alpelisib (ESCAT I) | Everolimus/Exemestan | in | 14 | 44 | 0.3 |
| 4 | PIK3CA | Alpelisib (ESCAT I) | Palbociclib/Anastrozol | in | 15 | 32 | 0.5 |
| 5 | FGFR1 | Everolimus | Trastuzumab/Eribulin | In | 4 | 8 | 0.5 |
| 6 | ERBB2 | Trastuzumab/Lapatinib (ESCAT I) | Trastuzumab-Emtansin | in | 21 | 25 | 0.8 |
| 7 | FGFR1 | Everolimus | Eribulin | in | 13 | 13 | 1 |
| 8 | PIK3CA | Everolimus | Trastuzumab/Pertuzumab | in | 69 | 55 | 1.3 |
| 9 | FGFR1 | Everolimus | Docetaxel/Pertuzumab/Trastuzumab | in | 13 | 9 | 1.4 |
| 10 | PIK3CA | Everolimus | Paclitaxel | in | 18 | 12 | 1.5 |
| 11 | CCND1 | Palbociclib | Carboplatin/Gemcitabine | in | 21 | 13 | 1.6 |
| 12 | PIK3CA | Alpelisib (ESCAT I) | Carboplatin/Gemcitabine | in | 15 | 9 | 1.7 |
| 13 | FGFR1 | Pazopanib | Cyclophosphamid/Methotrexat/Fluorouracil | off | 12 | 6 | 2 |
| 14 | FGFR1 | Pazopanib | Eribulin | off | 6 | 3 | 2 * |
| 15 | ERBB2 | Lapatinib (ESCAT II) | Epirubicin | in | 26 | 3 | 8.7 |
| 16 | p16, MYC | Pembrolizumab [33,34,35] | Cisplatin/5-Fluorouracil | off | 59 | 5 | 11.8 |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| MTB | Metastatic breast cancer |
| PD-L1 | Programmed death-ligand 1 |
| TNBC | Triple-negative breast cancer |
| PARP | Poly (ADP-ribose) polymerase |
| gBRCA | Germline BRCA |
| CTC | Circulating tumor cell |
| LoE | Levels of evidence |
| GR | Grade |
| AGO | Arbeitsgemeinschaft Gynäkologische Onkologie (German Gynecological Oncology Group) |
| MTB | Molecular Tumor Board |
| ESCAT | ESMO Scale of Clinical Actionability |
| PFS | Progression-free survival |
| PFSr | Progression-free survival ratio |
| HR | Hormone receptor |
| ER | Estrogen receptor |
| PR | Progesterone receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| NGS | Next-generation sequencing |
| MTA | Molecular targeted agents |
| OR | Overall response rate |
| SD | Stable disease |
| PR | Partial remission |
Appendix A
| # | Alteration Found | Treatment Recommendation | Follow-Up | |
| 1 | PIK3CA, ESR1 and TP53 mutations | PIK3CA-Gene: chr3: 178952085, Exon 21, c.3140A > G (NM_006218.3), p.His1047Arg | alpelisib | lost to follow up |
| ESR1-Gene: chr6: 152332832, Exon 6, c.1138G > C (NM_001122740.1), p.Glu380Gln | ||||
| TP53-Gene: chr17: 7579558, Exon 4, c.128delT (NM_000546.5), p.Leu43Ter | ||||
| 2 | FGFR1, AR and CCND1 amplifications | FGFR1-Gene: chr8: 38271444, CNV: 9.83 |
| not implemented |
| AR-Gene: chrX: 66776185, CNV: 8 | ||||
| CCND1-Gene: chr11: 69456941, CNV: 10.57 | ||||
| 3 | PIK3CA and ESR1 mutations; TBL1XR1-PIK3CA gene fusion | External analysis |
| implemented |
| 4 | PIK3CA mutation | PIK3CA-Gene: chr3: 178936091, Exon 10, c. 1633G>A (NM_006218.3), p.Glu545Lys | alpelisib | implemented |
| 5 | ERBB2 amplification | External analysis | HER2 inhibitor | not implemented |
| 6 | CCND1 amplification | CCND1-Gene: 11q13.3, chr11: 69456941, CNV: 9.3 |
| implemented |
| 7 | PIK3CA, PTEN mutations and AKT3 amplification | PIK3CA-Gene: chr3: 178936082, Exon 10, c. 1624G > A (NM_006218.3), p.Glu542Lys | mTOR inhibitor | implemented |
| PTEN-Gene: chr10: 89720803, Exon 8, c.955_956insA, (NM_000314.6 p.Thr319fs | ||||
| AKT3-Gene: 1q43q44, chr:1: 243662992, CNV: 5.9 | ||||
| 8 | FGFR1 and AKT2 amplifications; TP53 mutation | FGFR1-Gene: 8p11.23, chr8: 38271114, CNV: 15.33 | FGF1 inhibitor | not implemented |
| AKT2-Gene: 19q13.2, chr19: 40739755, CNV: 11.61 | ||||
| TP53-Gene: chr17: 7579320, Exon 4, c.365_366delTG (NM_000546.5), p.Val122fs | ||||
| 9 | ERBB2 mutation | ERBB2-Gene: Exon 19, chr17: 37880220, c.2264T > C (NM_004448.3), p.Leu755Ser | afatinib/neratinib | not implemented |
| 10 | PTEN deletion | PTEN-Gene: Exon 8, chr10: 89720798, c.955_958delACTT (NM_000314.4), p.Thr319Ter |
| not implemented |
| 11 | PIK3CA mutation | PIK3CA-Gene: chr3: 178936091, Exon 10, c. 1633G > A (NM_001127500.2), p.Glu545Lys | everolimus | not implemented |
| 12 | FGFR1, FGF19 and FGF3 mutations | FGFR1-Gene: 8p11.23, chr8: 38271114, CNV: 24.97 |
| implemented |
| FGF19-Gene: 11q13.3, chr11: 69513954, CNV: 19.73 | ||||
| FGF3-Gene: 11q13.3, chr11: 69624976, CNV: 12.97 | ||||
| 13 | MET mutation | MET-Gene: Exon 14, chr7: 116411990, c.3029C > T (NM_001127500.1), p.Thr1010Ile | crizotinib | not implemented |
| 14 | MYC, FGFR1 and CCND1 amplifications | MYC-Gene: chr8: 128748884, CNV: 18.8 | everolimus | implemented |
| FGFR1-Gene: chr8: 38271444, CNV: 20.13 | ||||
| CCND1-Gene: chr11: 69456941, CNV: 38.33 | ||||
| 15 | BRCA1 mutation; AR amplification | BRCA1 (external analysis)AR-Gene: chrX: 66776185, CNV: 7.87 |
| not implemented |
| 16 | PIK3CA mutation | PIK3CA-Gene: Exon21, chr3: 178952074, c. 3129G > A (NM_006218.2), p.Met1043Ile |
| not implemented |
| 17 | AKT2 amplification; SF3B1 mutation | AKT2-Gene: 19q13.2, chr19: 40739755, CNV: 5.44 | everolimus + hormone therapy | not implemented |
| SF3B1-Gene: chr2: 198266834, Exon 15, c.2098A > G (NM_012433.3), p.Lys700Glu | ||||
| 18 | PIK3CA mutation; MET amplification | PIK3CA-Gene: chr3: 178952085, Exon 21, c. 3140A > G (NM_006218.3), p.His1047Arg | crizotinib | not implemented |
| MET-Gene: 7q31.2, chr7: 116339592, CNV: 4.61 | ||||
| 19 | FGFR1 amplification | FGFR1-Gene: 8p11.23, chr8: 38271114, CNV: 9.6 |
| not implemented |
| 20 | ESR1 and PALB2 mutations; ESR1-CCDC170 fusion | ESR1-Gene: chr6: 152419923, Exon 9, c.1610A > C (NM_001122740.1), p.Tyr537Ser |
| not implemented |
| PALB2-Gene: chr16: 23641065, Exon 5, c.2409_2410insAC (NM_024675.3), p.Ser804fs | ||||
| ESR1-CCDC170 fusion: chr6: 151894309, t(6;6) (q25;q25), ESR1(Ex2)-CCDC170(Ex6) | ||||
| 21 | FGFR1 and MYC amplifications; TP53 mutation | FGFR1-Gene: 8p11.23, chr8: 38271114, CNV: 19.83 |
| not implemented |
| MYC-Gene: 8q24.21, chr8: 128748724, CNV: 12.17 | ||||
| TP53-Gene: chr17: 7578500, Exon 5, c.423_432delinsCA (NM_00546.5), p.Pro 142fs | ||||
| 22 | ERBB2 amplification | ERBB2-Gene: chr17: 37868125, CNV: 24.98 | lapatinib, trastuzumab emtansine and pertuzumab | not implemented |
| 23 | ARID1A and PIK3CA mutations | ARID1A-Gene: chr1: 27023060, Exon 1, c.166C > T (NM_006015.5), p.Gln56Ter | everolimus | implemented |
| PIK3CA-Gene: chr3: 178936091, Exon 10, c.1633G > A (NM_006218.3), p.Glu545Lys | ||||
| 24 | ESR1 mutation | ESR1-Gene: chr6: 152419926, Exon 9, c.1613A > G (NM_001122740.1), p.Asp538Gly | fulvestrant + everolimus | not implemented |
| 25 | FGFR1, CCND1, FGF19 and IGF1R amplifications; ATM mutation | FGFR1-Gene: 8p11.23, chr8: 38271114, CNV: 10.33 | mTOR inhibitor | implemented |
| CCND1-Gene: 11q13.3, chr11: 69455972, CNV: 7.54 | ||||
| FGF19-Gene: 11q13.3, chr11: 69513954, CNV: 7.95 | ||||
| IGF1R-Gene: 15q26.3, chr15: 99192814, CNV: 27.06 | ||||
| ATM-Gene: chr11: 108190701, Exon 44, c.6370_6371insT (NM_000051.3), p. Tyr2124fs | ||||
| 26 | TP53 mutation; FGFR1, CCND1, FGF19 und FGF3 amplifications | TP53-Gene: chr17: 7577121, Exon 8, c.817C > T (NM_000546.5), p.Arg273Cys | FGFR1 inhibitor | not implemented |
| FGFR1-Gene: 8p11.23, chr8: 38271114, CNV: 20.63 | ||||
| CCND1-Gene: 11q13.3, chr11: 69455972, CNV: 8.37 | ||||
| FGF19-Gene: 11q13.3, chr11: 69513954, 1 CNV: 0.93 | ||||
| FGF3-Gene: 11q13.3, chr11: 69624976, CNV: 15.77 | ||||
| 27 | PIK3CA, ERBB2, CDKN2A mutations | PIK3CA-Gene: chr3:178936091, Exon 10, c.1633G > A (NM_006218.3), p.Glu545Lys |
| not implemented |
| ERBB2-Gene: chr17: 37880219, Exon 19, c.2264T > C (NM_004448.3), p.Leu755Ser | ||||
| CDKN2A-Gene: chr9:21974748, Exon 1, c.79G > T (NM_001195132.1, p.Glu27Ter | ||||
| 28 | TPM3(7)-NTRK1(10) fusion | TPM3(7)-NTRK1(10)-Gene: Exon 7 (I), chr1: 154142875-chr1: 156844362 | trial (NCT02568267) | not implemented |
| 29 | MET mutation | MET-Gene: Exon 2, chr7: 116411990, c.3029C > T (NM_001127500.1), p.Thr1010Ile | cabozantinib | not implemented |
| 30 | KRAS and PIK3CA mutations | KRAS-Gene: chr12: 25398284, Exon 2, c.34G > C (NM_033360.3), p.Gly12Arg | peg-Doxorubicin/bevacizumab und temsirolimus/everolimus | not implemented |
| PIK3CA-Gene: chr3: 178936082, Exon 10, c.1624G > A (NM_006218.3), p.Glu542Lys | ||||
| PIK3CA-Gene: chr3: 178938934, Exon 14, c.2176G > A (NM_006218.3), p.Glu726Lys | ||||
| 31 | AR and PIK3CA mutations | AR-Gene: chrX: 66941751, Exon 6, c.2395C > G (NM_000044.3), p.Gln799Glu | everolimus | not implemented |
| PIK3CA-Gene: chr3: 178936091, Exon 10, c.1633G > A (NM_006218.3), p.Glu545Lys | ||||
| 32 | MET, CCND1, FGF19, FGF3 amplifications | MET-Gene: 7q31.2, chr7: 116339592, CNV: 4.66 | FGF1 inhibitor | implemented |
| CCND1-Gene: 11q13.3, chr11: 69455972, CNV: 20.87 | ||||
| FGF19-Gene: 11q13.3, chr11: 69513954, CNV: 18.61 | ||||
| FGF3-Gene: 11q13.3, chr11: 69624976, CNV: 18.49 | ||||
| 33 | FGFR1, CCND1, EGFR, PIK3CA und PDGFRA amplifications | External analysis | pazopanib | not implemented |
| 34 | PIK3CA mutation; ERBB2 amplification | External analysis |
| not implemented |
| 35 | ERBB2 mutation; CCNE1, AKT2, ERBB2 amplifications | ERBB2-Gene: chr17: 37868208, Exon 8, c.929C > A (NM_004448.3), p.Ser310Tyr | trastuzumab + lapatinib | implemented |
| CCNE1-Gene: 19q12, chr19: 30303882, CNV: 5.51 | ||||
| AKT2-Gene: 19q13.2, chr19: 40739755, CNV: 6.09 | ||||
| ERBB2-Gene: 17q12, chr17: 37868168, CNV: 8.46 | ||||
| 36 | ESR1 and PIK3CA mutations | ESR1-Gene: chr6: 152419922, Exon 9, c.1609T > A (NM_001122740.1), p.Tyr537Asn |
| not implemented |
| PIK3CA-Gene: chr3: 178936091, Exon 10, c.1633G > A (NM_006218.3), p.Glu545Lys | ||||
| 37 | p16 high expression and MYC amplification | MYC-Gene: chr8: 128748884, CNV: 5.61 | checkpoint inhibitors | implemented |
| 38 | AKT3 amplification and TP53 mutation | AKT3-Gene: 1q43q44, chr1: 243662992, CNV: 6.04 | MASTER trial | not implemented |
| TP53-Gene: chr17: 7578189, Exon 6, c.660T > A (NM_000546.5), p.Tyr220Ter | ||||
| 39 | AR amplification | AR-Gene: chrX: 66776185, CNV: 7.65 | AR inhibitors | not implemented |
| 40 | AKT1 mutation | AKT1-Gene: chr14: 105246551, Exon 3, c.49G > A (NM_001014431.1), p.Glu17Lys |
| not implemented |
| 41 | SLX4 mutation; FGFR1, CCND1, FGF19, FGFR3 amplifications | SLX4-Gene: chr16: 3640038, Exon 12, c.3601C > T (NM_032444.3), p.Gln1201Ter | pazopanib | implemented |
| FGFR1-Gene: 8p11.23, chr8: 38271114, CNV: 15.4 | ||||
| CCND1-Gene: 11q13.3, chr11: 69455972, CNV: 5.83 | ||||
| FGF19-Gene: 11q13.3, chr11: 69513954, CNV: 6.01 | ||||
| FGF3-Gene: 11q13.3, chr11: 69624976, CNV: 6.05 | ||||
| 42 | ESR1 mutation | ESR1-Gene: Exon 9, chr6: 152419919, c.1606_1608delCTCinsAAA (NM_001122740.1), p.Leu536Lys | fulvestrant + CDK4/6 inhibitors | not implemented |
| 43 | CCND1 and FGF19 amplifications; AKT1 mutation | CCND1-Gene: 11q13.3, chr11: 69455972, CNV: 9.13 |
| not implemented |
| FGF19-Gene: 11q13.3, chr11: 69513954, CNV: 9.99 | ||||
| AKT1-Gene: chr14:105246551, Exon 3, c.49G > A (NM_001014431.1), p.Glu17Lys | ||||
| 44 | PIK3CA and TP53 mutations | PIK3CA-Gene: chr3: 178952085, Exon 21, c.3140A > G (NM_006218.3), p.HIS1047Arg | alpelisib | implemented |
| TP53-Gene: chr17: 7577538, Exon 7, c.743G > A (NM_000546.5), p.Arg248Gln | ||||
| 45 | CCND1 and FGFR1 amplifications | External analysis |
| not implemented |
| 46 | PIK3CA and ERBB2 mutations; ERBB2 high expression | PIK3CA-Gene: chr3: 178927980, Exon 8, c.1258T > C (NM_006218.3), p.Cys420Arg |
| implemented |
| ERBB2-Gene: chr17: 37881000, Exon 20, c.2329G > T (NM_004448.3), p.Val777Leu | ||||
| 47 | FGFR1 amplification | FGFR1-Gene: 8p11.23p11.22, chr8: 38271444, CNV: 6.61 | everolimus + hormone therapy | implemented |
| 48 | CCND1 amplification | CCND1-Gene: 11q13.3, chr11: 69456942, CNV: 5.48 | exemestane + everolimus | not implemented |
| 49 | CCND1 and FGFR1 amplifications | CCND1-Gene: 11q13.3, chr11: 69456941, CNV: 20.18 |
| implemented |
| FGFR1-Gene: 8p11.23p11.22, chr8: 38271444, CNV: 28.5 | ||||
References
- Cancer Today. Breast Cancer Incidence and Mortality. 2018. Available online: https://gco.iarc.fr/today/online-analysis-table?v=2018&mode=cancer&mode_population=continents&population=900&populations=900&key=asr&sex=2&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&group_cancer=1&include_nmsc=1&include_nmsc_other=1 (accessed on 10 March 2021).
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol. Biomarkers Prev. 2015, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Cancer Stat Facts: Female Breast Cancer. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 22 March 2021).
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Lopez, B.A.; Barrios, C.; Bergh, J.; et al. 4th ESO–ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4). Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef]
- American Cancer Society. Survival Rates for Breast Cancer. 2020. Available online: https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html (accessed on 10 April 2021).
- Lu, J.; Steeg, P.S.; Price, J.E.; Krishnamurthy, S.; Mani, S.A.; Reuben, J.; Cristofanilli, M.; Dontu, G.; Bidaut, L.; Valero, V.; et al. Breast Cancer Metastasis: Challenges and Opportunities. Cancer Res. 2009, 69, 4951–4953. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, N.; Düring, M.; Rasmussen, B.B.; Mouridsen, H.; Kroman, N. Prognostic effect of estrogen receptor status across age in primary breast cancer. Int. J. Cancer 2007, 122, 1089–1094. [Google Scholar] [CrossRef]
- Sopik, V.; Sun, P.; Narod, S.A. The prognostic effect of estrogen receptor status differs for younger versus older breast cancer patients. Breast Cancer Res. Treat. 2017, 165, 391–402. [Google Scholar] [CrossRef]
- Yu, K.-D.; Wu, J.; Shen, Z.-Z.; Shao, Z.-M. Hazard of Breast Cancer-Specific Mortality among Women with Estrogen Receptor-Positive Breast Cancer after Five Years from Diagnosis: Implication for Extended Endocrine Therapy. J. Clin. Endocrinol. Metab. 2012, 97, E2201–E2209. [Google Scholar] [CrossRef]
- Jayasekara, H.; MacInnis, R.J.; Chamberlain, J.A.; Dite, G.S.; Leoce, N.M.; Dowty, J.G.; Bickerstaffe, A.; Win, A.K.; Milne, R.L.; Giles, G.G.; et al. Mortality after breast cancer as a function of time since diagnosis by estrogen receptor status and age at diagnosis. Int. J. Cancer 2019, 145, 3207–3217. [Google Scholar] [CrossRef]
- Garraway, L.A. Genomics-Driven Oncology: Framework for an Emerging Paradigm. J. Clin. Oncol. 2013, 31, 1806–1814. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Chuang, J.C.; Neal, J.W. Crizotinib as first line therapy for advanced ALK-positive non-small cell lung cancers. Transl. Lung Cancer Res. 2015, 4, 639–641. [Google Scholar]
- Chapman, P.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Jungsil CLEOPATRA Study Group; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, Trastuzumab, and Docetaxel in HER2-Positive Metastatic Breast Cancer. New Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Kim, S.-B.; González-Martín, A.; LoRusso, P.M.; Ferrero, J.-M.; Smitt, M.; Yu, R.; Leung, A.C.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. New Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Dawood, S.; Broglio, K.; Buzdar, A.U.; Hortobagyi, G.N.; Giordano, S.H. Prognosis of Women with Metastatic Breast Cancer by HER2 Status and Trastuzumab Treatment: An Institutional-Based Review. J. Clin. Oncol. 2010, 28, 92–98. [Google Scholar] [CrossRef]
- Smyth, L.; Saura, C.; Piha-Paul, S.; Lu, J.; Mayer, I.; Brufksy, A.; Spanggaard, I.; Arnedos, M.; Cutler, R.; Hyman, D. Update on the phase II SUMMIT trial: Neratinib + fulvestrant for HER2-mutant, HR-positive, metastatic breast cancer. Ann. Oncol. 2019, 30, iii10–iii11. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. New Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Li, X.; Warner, J.L. A Review of Precision Oncology Knowledgebases for Determining the Clinical Actionability of Genetic Variants. Front. Cell Dev. Biol. 2020, 8, 48. [Google Scholar] [CrossRef]
- Condorelli, R.; Mosele, F.; Verret, B.; Bachelot, T.; Bedard, P.; Cortes, J.; Hyman, D.; Juric, D.; Krop, I.; Bieche, I.; et al. Genomic alterations in breast cancer: Level of evidence for actionability according to ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2019, 30, 365–373. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.; Barlesi, F.; Lolkema, M.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Stephenson, J.J.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef]
- Schwartz, L.H.; Litière, S.; De Vries, E.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1—Update and clarification: From the RECIST committee. Eur. J. Cancer 2016, 62, 132–137. [Google Scholar] [CrossRef]
- Taghizadeh, H.; Mader, R.M.; Müllauer, L.; Aust, S.; Polterauer, S.; Kölbl, H.; Seebacher, V.; Grimm, C.; Reinthaller, A.; Prager, G.W. Molecular Guided Treatments in Gynecologic Oncology: Analysis of a Real-World Precision Cancer Medicine Platform. Oncol. 2020, 25, e1060–e1069. [Google Scholar] [CrossRef]
- Drago, J.Z.; Formisano, L.; Juric, D.; Niemierko, A.; Servetto, A.; Wander, S.A.; Spring, L.M.; Vidula, N.; Younger, J.; Peppercorn, J.; et al. FGFR1 Amplification Mediates Endocrine Resistance but Retains TORC Sensitivity in Metastatic Hormone Receptor–Positive (HR+) Breast Cancer. Clin. Cancer Res. 2019, 25, 6443–6451. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Chen, D.; Piccart, M.; Rugo, H.S.; Burris, H.A., 3rd; Pritchard, K.I.; Campone, M.; Noguchi, S.; Perez, A.T.; Deleu, I.; et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J. Clin Oncol. 2016, 34, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Mai, S. c-MYC-Induced Genomic Instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef] [PubMed]
- Kwei, K.A.; Kung, Y.; Salari, K.; Holcomb, I.N.; Pollack, J.R. Genomic instability in breast cancer: Pathogenesis and clinical implications. Mol. Oncol. 2010, 4, 255–266. [Google Scholar] [CrossRef]
- Yaghmour, G.; Pandey, M.; Ireland, C.; Patel, K.; Nunnery, S.; Powell, D.; Baum, S.; Wiedower, E.; Schwartzberg, L.S.; Martin, M.G. Role of Genomic Instability in Immunotherapy with Checkpoint Inhibitors. Anticancer. Res. 2016, 36, 4033–4038. [Google Scholar] [CrossRef]
- Mock, A.E.; Heilig, C.; Kreutzfeldt, S.; Huebschmann, D.; Heining, C.; Schröck, E.; Brors, B.; Stenzinger, A.; Jäger, D.; Schlenk, R.; et al. Community-driven development of a modified progression-free survival ratio for precision oncology. ESMO Open 2019, 4, e000583. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Hoefflin, R.; Geißler, A.-L.; Fritsch, R.; Claus, R.; Wehrle, J.; Metzger, P.; Reiser, M.; Mehmed, L.; Fauth, L.; Heiland, D.H.; et al. Personalized Clinical Decision Making Through Implementation of a Molecular Tumor Board: A German Single-Center Experience. JCO Precis. Oncol. 2018, 2, 1–16. [Google Scholar] [CrossRef]
- Hempel, D.; Ebner, F.; Garg, A.; Trepotec, Z.; Both, A.; Stein, W.; Gaumann, A.; Güttler, L.; Janni, W.; DeGregorio, A.; et al. Real world data analysis of next generation sequencing and protein expression in metastatic breast cancer patients. Sci. Rep. 2020, 10, 10459. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Bachelot, T.; Commo, F.; Campone, M.; Arnedos, M.; Dieras, V.; Lacroix-Triki, M.; Lacroix, L.; Cohen, P.; Gentien, D.; et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: A multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 2014, 15, 267–274. [Google Scholar] [CrossRef]
- Van Geelen, C.T.; Savas, P.; Teo, Z.L.; Luen, S.J.; Weng, C.-F.; Ko, Y.-A.; Kuykhoven, K.S.; Caramia, F.; Salgado, R.; Francis, P.A.; et al. Clinical implications of prospective genomic profiling of metastatic breast cancer patients. Breast Cancer Res. 2020, 22, 91. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Kolberg-Liedtke, C.; Wuerstlein, R.; Gluz, O.; Heitz, F.; Freudenberger, M.; Bensmann, E.; du Bois, A.; Nitz, U.; Pelz, E.; Warm, M.; et al. Phenotype Discordance between Primary Tumor and Metastasis Impacts Metastasis Site and Outcome: Results of WSG-DETECT-PriMet. Breast Care 2020, 1–9. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R. Comprehensive molecular portraits of human breast tumours. Nature. 2012, 490, 61–70. [Google Scholar]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef]
- Pagès, A.; Foulon, S.; Zou, Z.; Lacroix, L.; Lemare, F.; De Baère, T.; Massard, C.; Soria, J.-C.; Bonastre, J. The cost of molecular-guided therapy in oncology: A prospective cost study alongside the MOSCATO trial. Genet. Med. 2016, 19, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.A.; Schwaederlé, M.; Scur, M.D.; Boles, S.G.; Helsten, T.; Subramanian, R.; Schwab, R.B.; Kurzrock, R. Breast Cancer Experience of the Molecular Tumor Board at the University of California, San Diego Moores Cancer Center. J. Oncol. Pr. 2015, 11, 442–449. [Google Scholar] [CrossRef]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Fanta, P.T.; Boles, S.G.; Daniels, G.A.; Bazhenova, L.A.; Subramanian, R.; Coutinho, A.C.; Ojeda-Fournier, H.; et al. Molecular Tumor Board: The University of California San Diego Moores Cancer Center Experience. Oncol. 2014, 19, 631–636. [Google Scholar] [CrossRef]
- Hsu, J.C.; Lu, C.Y. Longitudinal trends in use and costs of targeted therapies for common cancers in Taiwan: A retrospective observational study. BMJ Open 2016, 6, e011322. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, J.; LaVange, L.M. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. New Engl. J. Med. 2017, 377, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, D.L.; van Herpen, C.M.L.; van Laarhoven, H.W.M.; Smit, E.F.; Groen, H.J.M.; Willems, S.M.; Nederlof, P.M.; Langenberg, M.H.; Cuppen, E.; Sleijfer, S.; et al. Molecular Tumor Boards: Current practice and future needs. Ann. Oncol. 2017, 28, 3070–3075. [Google Scholar] [CrossRef]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.; André, F.; Barrios, C.; Bergh, J.; Bhattacharyya, G.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef] [PubMed]
- Hirshfield, K.M.; Tolkunov, D.; Zhong, H.; Ali, S.M.; Stein, M.N.; Murphy, S.; Vig, H.; Vazquez, A.; Glod, J.; Moss, R.A.; et al. Clinical Actionability of Comprehensive Genomic Profiling for Management of Rare or Refractory Cancers. Oncol. 2016, 21, 1315–1325. [Google Scholar] [CrossRef]
- Sultova, E.; Westphalen, C.B.; Jung, A.; Kumbrink, J.; Kirchner, T.; Mayr, D.; Rudelius, M.; Ormanns, S.; Heinemann, V.; Metzeler, K.H.; et al. NGS-guided precision oncology in metastatic breast and gynecological cancer: First experiences at the CCC Munich LMU. Arch. Gynecol. Obstet. 2020, 1–15. [Google Scholar] [CrossRef]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a026617. [Google Scholar] [CrossRef]






| Author/Study | Tumor Entity | Enrolled Patients (n =) | MP Patients | Actionable Alterations | Implemented Therapies-n (% of Enrolled) | Results |
|---|---|---|---|---|---|---|
| André et al. (SAFIR01/UNICANCER) | breast cancer | 423 | 299 (71%) | 195 (46%) | 55 (13%) | ORR: 4 patients had a PR and 9 had SD > 16 weeks (3% of all patients) |
| Parker et al. | breast cancer | 43 | 43 (100%) | 40 (93%) | 17 (40%) | 7 patients (16% of all patients) achieved SD or PR |
| Van Geelen et al. | breast cancer | 322 | 234 (72%) | 74 (23%) | No data | No data about implementation rate and outcome |
| Genomic Alterations | Prevalence |
|---|---|
| ESCAT Level I | |
| BRCA1/2 germline mutations | 4% |
| ERBB2 amplifications | 15–20% |
| Microsatellite instability-high | 1% |
| PIK3CA hotspot mutations | 30–40% |
| ESCAT Level II | |
| AKT1E17K mutations | 5% |
| ERBB2 hotspot mutations | 4% |
| ESR1 mutations | 10% |
| ESCAT Level III | |
| BRCA1/2 somatic mutations | 3% |
| ERBB3 mutations | 2% |
| MDM2 amplifications | 1% |
| Patient Characteristics | n = |
|---|---|
| Median age | 52 (range 30–82) |
| Number of metastatic sites at time of presentation | |
| 1 | 25 |
| 2 | 39 |
| 3 | 20 |
| >3 | 16 |
| Metastatic sites | |
| visceral | 87 |
| bone | 62 |
| brain | 21 |
| cutaneous | 11 |
| Number of previous therapies | |
| 1 | 6 |
| 2 | 26 |
| 3 | 13 |
| >3 | 55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultova, E.; Westphalen, C.B.; Jung, A.; Kumbrink, J.; Kirchner, T.; Mayr, D.; Rudelius, M.; Ormanns, S.; Heinemann, V.; Metzeler, K.H.; et al. Implementation of Precision Oncology for Patients with Metastatic Breast Cancer in an Interdisciplinary MTB Setting. Diagnostics 2021, 11, 733. https://doi.org/10.3390/diagnostics11040733
Sultova E, Westphalen CB, Jung A, Kumbrink J, Kirchner T, Mayr D, Rudelius M, Ormanns S, Heinemann V, Metzeler KH, et al. Implementation of Precision Oncology for Patients with Metastatic Breast Cancer in an Interdisciplinary MTB Setting. Diagnostics. 2021; 11(4):733. https://doi.org/10.3390/diagnostics11040733
Chicago/Turabian StyleSultova, Elena, C. Benedikt Westphalen, Andreas Jung, Joerg Kumbrink, Thomas Kirchner, Doris Mayr, Martina Rudelius, Steffen Ormanns, Volker Heinemann, Klaus H. Metzeler, and et al. 2021. "Implementation of Precision Oncology for Patients with Metastatic Breast Cancer in an Interdisciplinary MTB Setting" Diagnostics 11, no. 4: 733. https://doi.org/10.3390/diagnostics11040733
APA StyleSultova, E., Westphalen, C. B., Jung, A., Kumbrink, J., Kirchner, T., Mayr, D., Rudelius, M., Ormanns, S., Heinemann, V., Metzeler, K. H., Greif, P. A., Hester, A., Mahner, S., Harbeck, N., & Wuerstlein, R. (2021). Implementation of Precision Oncology for Patients with Metastatic Breast Cancer in an Interdisciplinary MTB Setting. Diagnostics, 11(4), 733. https://doi.org/10.3390/diagnostics11040733