
Non-Alcoholic Fatty Liver Disease and Cardiovascular Comorbidities: Pathophysiological Links, Diagnosis, and Therapeutic Management


 ,
,  ,
,  ,
,  ,
, 

Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
- >5% macrovesicular steatosis;
- presence of inflammation;
- ballooning hepatocytes with predominantly centro-lobular distribution [9].
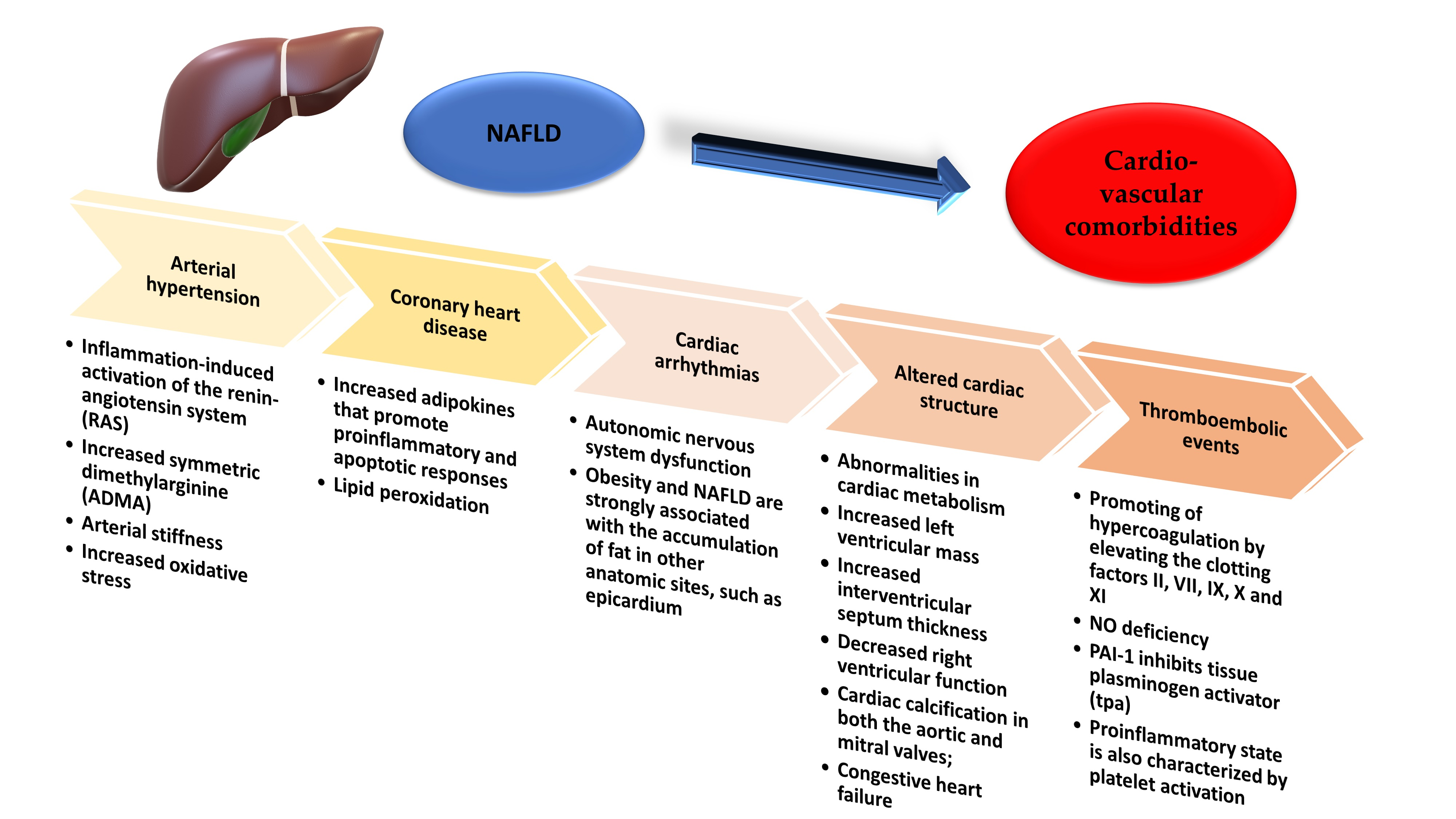
2. Non-Alcoholic Fatty Liver Disease and Arterial Hypertension
2.1. Systemic Inflammation
2.2. Insulin Resistance
2.3. Increased Vasoconstriction and Decreased Vasodilation
2.4. Arterial Stiffness
2.5. Increased Oxidative Stress
2.6. Gut Dysbiosis
2.7. Genetic and Epigenetic Modifications
3. Non-Alcoholic Fatty Liver Disease and Coronary Heart Disease
3.1. Insulin Resistance
3.2. Adipokines
3.3. Oxidative Stress
3.4. Lipid Peroxidation and Apoptosis
4. Non-Alcoholic Fatty Liver Disease and Cardiac Arrhythmias
4.1. Atrial Fibrillation
4.2. Ventricular Arrhythmias, Bundle Branch, and Atrioventricular Blocks
4.3. Mechanisms Behind Non-Alcoholic Fatty Liver Disease and Cardiac Arrhythmias
5. Non-Alcoholic Fatty Liver Disease and Altered Cardiac Structure
- abnormalities in cardiac metabolism;
- increased left ventricular mass;
- increased interventricular septum thickness;
- diastolic cardiac dysfunction;
- left atrium enlargement or impaired left atrium deformation;
- decreased right ventricular function;
- aortic and mitral valves calcification;
- congestive heart failure [111].
5.1. Abnormalities in Heart Metabolism
5.2. Increased Left Ventricular Mass and Interventricular Septum Thickness
5.3. Decreased Right Ventricular Function
5.4. Diastolic Cardiac Dysfunction and Risk of Congestive Heart Failure
6. Non-Alcoholic Fatty Liver Disease and Stroke
7. Non-Alcoholic Fatty Liver Disease and Thromboembolic Events
8. Therapeutic Management
8.1. Lifestyle Management and Dyslipidemia Treatment
8.2. Diabetes Mellitus Treatment
8.2.1. Pioglitazone
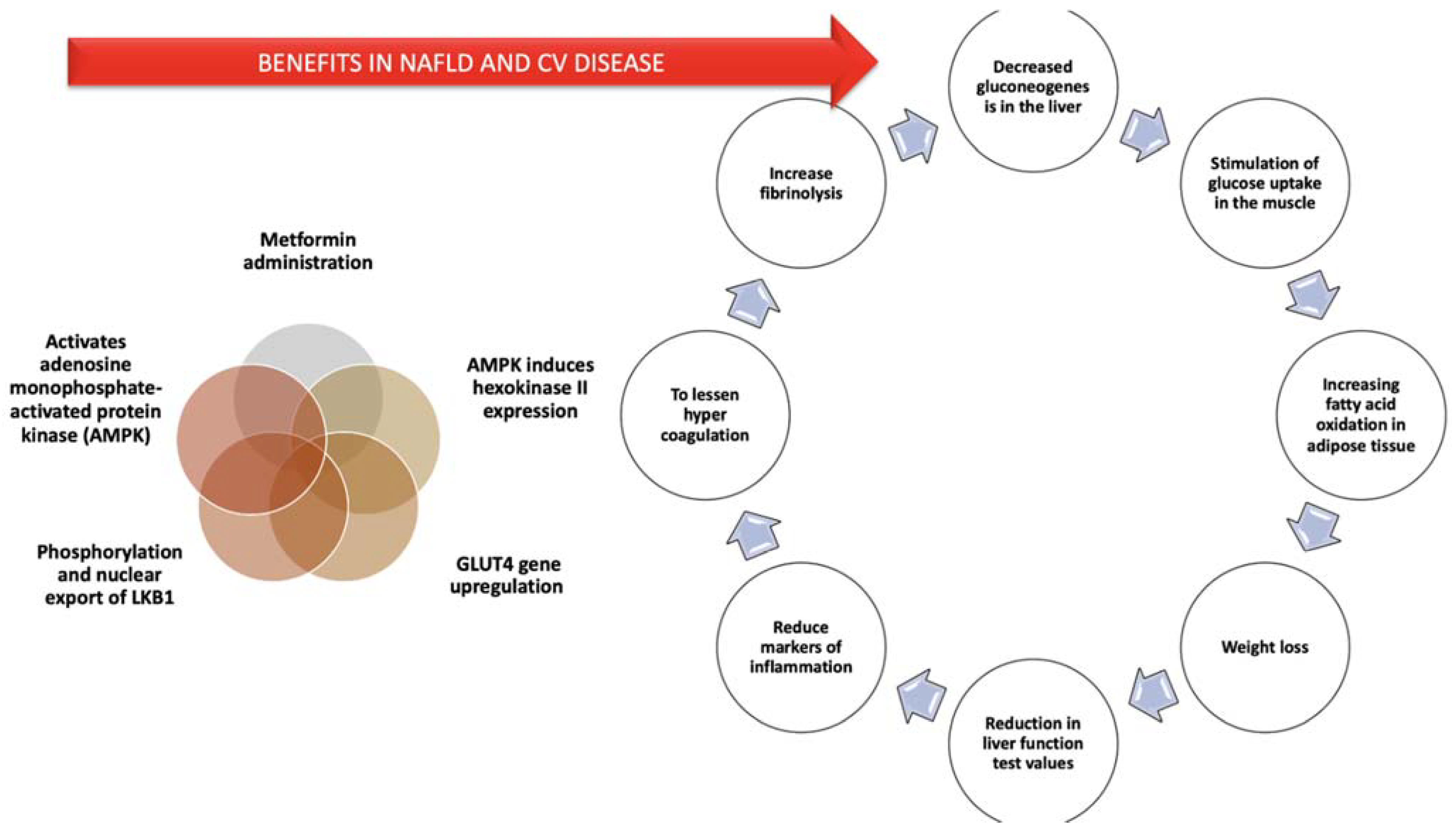
8.2.2. Metformin
8.2.3. Newer Antihyperglycemic Agents
8.3. Hypertension Treatment
8.3.1. Renin-Angiotensin-Aldosterone System Inhibitors
8.3.2. Probiotics and Prebiotics
8.4. Other Drugs That Might Have a Benefit in Non-Alcoholic Fatty Liver Disease
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jonathan, C.C.; Jay, D.H.; Helen, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 24, 1519–1523. [Google Scholar]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver. Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Z.; Hollis-Hansen, K.; Wan, X.Y.; Fei, S.J.; Pang, X.L.; Meng, F.D.; Yu, C.H.; Li, Y.M. Clinical Guidelines of Non-Alcoholic Fatty Liver Disease: A Systematic Review. World J. Gastroenterol. 2016, 22, 8226–8233. [Google Scholar] [CrossRef] [PubMed]
- Saverymuttu, S.H.; Joseph, A.E.; Maxwell, J.D. Ultrasound Scanning in the Detection of Hepatic Fibrosis and Steatosis. Br. Med. J. Clin. Res. 1986, 292, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.L.; Hamilton, G.; Patel, N.; O’Dwyer, R.; Dore, C.J.; Goldin, R.D.; Bell, J.D.; Taylor-Robinson, S.D. Hepatic Triglyceride Content and Its Relation to Body Adiposity: A Magnetic Resonance Imaging and Proton Magnetic Resonance Spectroscopy Study. Gut 2005, 54, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Desai, A.; Hamilton, G.; Wolfson, T.; Gamst, A.; Lam, J.; Clark, L.; Hooker, J.; Chavez, T.; Ang, B.D.; et al. Accuracy of MR Imaging Estimated Proton Density Fat Fraction for Classification of Dichotomized Histologic Steatosis Grades in Nonalcoholic Fatty Liver Disease. Radiology 2015, 274, 416–425. [Google Scholar] [CrossRef]
- Yoshio, S.; Atsushi, N.; Yoshito, I. Limitations of Liver Biopsy and Non-Invasive Diagnostic Tests for the Diagnosis of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. World J. Gastroenterol. 2014, 20, 475–485. [Google Scholar]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic Steatohepatitis: A Proposal for Grading and Staging the Histological Lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Neuman, M.G.; Malnick, S.; Maor, Y.; Nanau, R.M.; Melzer, E.; Ferenci, P.; Seitz, H.K.; Mueller, S.; Mell, H.; Samuel, D.; et al. Alcoholic Liver Disease: Clinical and Translational Research. Exp. Mol. Pathol. 2015, 99, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Jeon, W.K.; Kim, S.H.; Kim, H.J.; Park, D.I.; Cho, Y.K.; Sung, I.K.; Sohn, C.I.; Keum, D.K.; Kim, B.I. Prevalence and Risk Factors of Non-Alcoholic Fatty Liver Disease among Korean Adults. J. Gastroenterol. Hepatol. 2006, 21, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Nat Rev Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar]
- Yan-Ci, Z.; Guo-Jun, Z.; Ze, C.; Zhi-Gang, S.; Jingjing, C.; Hongliang, L. Nonalcoholic Fatty Liver Disease An Emerging Driver of Hypertension. Hypertension 2020, 75, 275–284. [Google Scholar]
- Catena, C.; Bernardi, S.; Sabato, N.; Grillo, A.; Ermani, M.; Sechi, L.A.; Fabris, B.; Carretta, R.; Fallo, F. Ambulatory Arterial Stiffness Indices and Nonalcoholic Fatty Liver Disease in Essential Hypertension. Nutr. Metab. Cardiovasc. Dis. 2012, 23, 389–393. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lo, H.M.; Chen, J.D. Sonographic Fatty Liver, Overweight and Ischemic Heart Disease. World J. Gastroenterol. 2005, 11, 4838. [Google Scholar] [CrossRef] [PubMed]
- Schindhelm, R.K.; Dekker, J.M.; Nijpels, G.; Bouter, L.M.; Stehouwer, C.D.A.; Heine, R.J.; Diamant, M. Alanine Aminotransferase Predicts Coronary Heart Disease Events: A 10-Year Follow-up of the Hoorn Study. Atherosclerosis 2007, 191, 391–396. [Google Scholar] [CrossRef]
- Sinn, D.H.; Gwak, G.Y.; Park, H.N.; Kim, J.E.; Min, Y.W.; Kim, K.M.; Kim, Y.J.; Choi, M.S.; Lee, J.H.; Koh, K.C.; et al. Ultrasonographically Detected Non-Alcoholic Fatty Liver Disease Is an Independent Predictor for Identifying Patients with Insulin Resistance in Non-Obese, Non-Diabetic Middle-Aged Asian Adults. Am. J. Gastroenterol. 2012, 107, 561–567. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Bo, S.; Uberti, S.; Biroli, G.; Pagano, G.; Cassader, M. Should Nonalcoholic Fatty Liver Disease Be Included in the Definition of Metabolic Syndrome? A Cross-Sectional Comparison with Adult Treatment Panel III Criteria in Nonobese Nondiabetic Subjects. Diabetes Care 2008, 31, 562–568. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Damås, J.K.; Konopski, Z.; Løberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjøro, K.; Aukrust, P. Systemic Inflammation in Nonalcoholic Fatty Liver Disease Is Characterized by Elevated Levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef]
- Stumpf, C.; Auer, C.; Yilmaz, A.; Lewczuk, P.; Klinghammer, L.; Schneider, M.; Daniel, W.G.; Schmieder, R.E.; Garlichs, C.D. Serum Levels of the Th1 Chemoattractant Interferon-γ-inducible Protein (IP) 10 Are Elevated in Patients with Essential Hypertension. Hypertens. Res. 2011, 34, 484–488. [Google Scholar] [CrossRef][Green Version]
- Bai, L.; Li, H.I. Immune Regulatory Networks in Hepatic Lipid Metabolism. J. Mol. Med. Berl. 2019, 97, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking Nonalcoholic Fatty Liver Disease and Insulin Resistance. Nat. Rev. Endocrinol. 2017, 13, 509. [Google Scholar] [CrossRef]
- Nunes, K.P.; de Oliveira, A.A.; Mowry, F.E.; Biancardi, V.C. Targeting Toll-like Receptor 4 Signalling Pathways: Can Therapeutics Pay the Toll for Hypertension? Br. J. Pharmacol. 2019, 176, 1864–1879. [Google Scholar] [CrossRef] [PubMed]
- Sinn, D.H.; Kang, D.; Jang, H.R.; Gu, S.; Cho, S.J.; Paik, S.W.; Ryu, S.; Chang, Y.; Lazo, M.; Guallar, E.; et al. Development of Chronic Kidney Disease in Patients with Non-Alcoholic Fatty Liver Disease: A Cohort Study. J. Hepatol. 2017, 67, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Häring, H.U. The Impact of Insulin Resistance on the Kidney and Vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737. [Google Scholar] [CrossRef]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Landsberg, L.; Young, J.B. Insulin Mediated Glucose Metabolism in the Relationship between Dietary Intake and Sympathetic Nervous System Activity. Int. J. Obes. 1985, 9, 63–68. [Google Scholar]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed Hepatic Bile Acid Signalling despite Elevated Production of Primary and Secondary Bile Acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Edmison, J.M.; Dasarathy, S.; Bennett, C.; Lopez, R.; Kalhan, S.C. Plasma levels of asymmetric dimethylarginine (ADMA) in patients with biopsy-proven non-alcoholic fatty liver disease. Metabolism 2011, 60, 776–781. [Google Scholar] [CrossRef]
- Serg, M.; Kampus, P.; Kals, J.; Zagura, M.; Muda, P.; Tuomainen, T.P.; Zilmer, K.; Salum, E.; Zilmer, M.; Eha, J. Association between Asymmetric Dimethylarginine and Indices of Vascular Function in Patients with Essential Hypertension. Blood Press 2011, 20, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Mondon, C.E.; Cooke, J.P. Insulin Resistance: Potential Role of the Endogenous Nitric Oxide Synthase Inhibitor ADMA. Vasc. Med. 2005, 10 (Suppl. 1), S35–S43. [Google Scholar] [CrossRef]
- Abbasi, F.; Asagmi, T.; Cooke, J.P.; Lamendola, C.; McLaughlin, T.; Reaven, G.M.; Stuehlinger, M.; Tsao, P.S. Plasma Concentrations of Asymmetric Dimethylarginine Are Increased in Patients with Type 2 Diabetes Mellitus. Am. J. Cardiol. 2001, 88, 1201–1203. [Google Scholar] [CrossRef]
- Villela-Nogueira, C.A.; Leite, N.C.; Cardoso, C.R.L.; Salles, G.F. NAFLD and Increased Aortic Stiffness: Parallel or Common Physiopathological Mechanisms? World J. Gastroenterol. 2014, 14, 8377–8392. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.P.; Baugh, R.; Wilson, C.A.; Burns, J. Age Related Changes in the Tunica Media of the Vertebral Artery: Implications for the Assessment of Vessels Injured by Trauma. J. Clin. Pathol. 2001, 54, 139–145. [Google Scholar] [CrossRef]
- Nickenig, G.; Roling, J.; Strehlow, K.; Schnabel, P.; Bohm, M. Insulin Induces Upregulation of Vascular AT1 Receptor Gene Expression by Posttranscriptional Mechanisms. Circulation 1998, 8, 2453–2460. [Google Scholar] [CrossRef] [PubMed]
- Jesmin, S.; Sakuma, I.; Salah-Eldin, A.; Nonomura, K.; Hattori, Y.; Kitabatake, A. Diminished Penile Expression of Vascular Endothelial Growth Factor and Its Receptors at the Insulin-Resistant Stage of a Type II Diabetic Rat Model: A Possible Cause for Erectile Dysfunction in Diabetes. J. Mol. Endocrinol. 2003, 31, 401–418. [Google Scholar] [CrossRef]
- Rizzoni, D.; Porteri, E.; Guelfi, D.; Muiesan, M.L.; Valentini, U.; Cimino, A.; Girelli, A.; Rodella, L.; Bianchi, R.; Sleiman, I.; et al. Structural Alterations in Subcutaneous Small Arteries of Normotensive and Hypertensive Patients with Non-Insulin-Dependent Diabetes Mellitus. Circulation 2001, 103, 1238–1244. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Manesis, E.; Baou, K.; Papatheodoridis, G.; Koskinas, J.; Tiniakos, D.; Aznaouridis, K.; Archimandritis, A.; Stefanadis, C. Increased Arterial Stiffness and Impaired Endothelial Function in Nonalcoholic Fatty Liver Disease: A Pilot Study. Am. J. Hypertens. 2010, 23, 1183–1189. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, N.H.; Kim, B.S.; Kang, J.H. The Association between Nonalcoholic Fatty Liver Disease, Metabolic Syndrome and Arterial Stiffness in Nondiabetic, Nonhypertensive Individuals. Cardiology 2012, 123, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Bi, Y.; Xu, M.; Ma, Z.; Xu, Y.; Wang, T.; Li, M.; Liu, Y.; Lu, J.; Chen, Y.; et al. Nonalcoholic Fatty Liver Disease Is Associated with Atherosclerosis in Middle-Aged and Elderly Chinese. Arter. Thromb. Vasc. Biol. 2012, 32, 2321–2326. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shim, J.Y.; Moon, B.S.; Shin, Y.H.; Jung, D.H.; Lee, J.H.; Lee, H.R. The Relationship between Arterial Stiffness and Nonalcoholic Fatty Liver Disease. Dig. Sci. 2012, 57, 196–203. [Google Scholar] [CrossRef]
- Ozturk, A.K.; Uygun, A.A.; Guler, B.A.K.; Demirci, A.H.; Ozdemir, C.C.; Cakir, B.M.; Sakin, A.Y.S.; Turker, D.T.; Sari, E.S.; Demirbas, B.S.; et al. Nonalcoholic Fatty Liver Disease Is an Independent Risk Factor for Atherosclerosis in Young Adult Men. Atherosclerosis 2015, 240, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Khera, R.; Corrales-Medina, V.F.; Townsend, R.R.; Chirinos, J.A. Inflammation and Arterial Stiffness in Humans. Atherosclerosis 2014, 237, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Farzanegi, P.; Amir, D.; Ebrahimpoor, Z.; Mahdieh, A.; Azarbayjani, M.A. Mechanisms of Beneficial Effects of Exercise Training on Non-Alcoholic Fatty Liver Disease (NAFLD): Roles of Oxidative Stress and Inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003. [Google Scholar] [CrossRef]
- Colagar, A.H.; Marzony, E.T. Ascorbic Acid in Human Seminal Plasma: Determination and Its Relationship to Sperm Quality. J. Clin. Biochem. Nutr. 2009, 45, 144–149. [Google Scholar] [CrossRef]
- Yu, Y.; Cai, J.; She, Z.; Li, H. Insights into the Epidemiology, Pathogenesis, and Therapeutics of Nonalcoholic Fatty Liver Diseases. Adv. Sci. Weinh. 2019, 6, 1801585. [Google Scholar] [CrossRef] [PubMed]
- Distrutti, E.; Mencarelli, A.; Santucci, L.; Renga, B.; Orlandi, S.; Donini, A.; Shah, V.; Fiorucci, S. The Methionine Connection: Homocysteine and Hydrogen Sulfide Exert Opposite Effects on Hepatic Microcirculation in Rats. Hepatology 2008, 47, 659–667. [Google Scholar] [CrossRef]
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative Stress: New Insights on the Association of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.; Yao, T.; Li, D.; Wang, Y.; Li, Y.; Wu, S.; Cai, J. Homocysteine; as a Risk Factor for Hypertension: A 2-Year Follow-up Study. PLoS ONE 2014, 9, e108223. [Google Scholar] [CrossRef]
- Björkholm, B.; Bok, C.M.; Lundin, A.; Rafter, J.; Hibberd, M.L.; Pettersson, S. Intestinal Microbiota Regulate Xenobiotic Metabolism in the Liver. PLoS ONE 2009, 4, e6958. [Google Scholar] [CrossRef]
- Safari, Z.; Gérard, P. The Links between the Gut Microbiome and Non Alcoholic Fatty Liver Disease (NAFLD). Cell. Mol. Life Sci. 2019, 76, 1541–1558. [Google Scholar] [CrossRef]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond Gut Feelings; How the Gut Microbiota Regulates Blood Pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Qu, X.; Li, J.; Wang, X.; Bai, Y.; Cao, Q.; Ma, L.; Zhou, X.; Zhu, A.W.; Liu, B.W.; et al. Associations between Polymorphisms of the ADIPOQ Gene and Hypertension Risk: A Systematic and Meta-Analysis. Sci. Rep. 2017, 7, 41683. [Google Scholar] [CrossRef]
- Zhu, W.; Cheng, K.K.; Vanhoutte, P.M.; Lam, K.S.; Xu, A. Vascular Effects of Adiponectin: Molecular Mechanisms and Potential Therapeutic Intervention. Clin. Sci. 2008, 114, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Saba, F.; Cassader, M.; Paschetta, E.; De Michieli, F.; Pinach, S.; Framarin, L.; Berrutti, M.; Leone, N.; Parente, R.; et al. Angiotensin II Type 1 Receptor Rs5186 Gene Variant Predicts Incident NAFLD and Associated Hypertension: Role of Dietary Fat-Induced pro-Inflammatory Cell Activation. Am. J. Gastroenterol. 2019, 114, 607–619. [Google Scholar] [CrossRef]
- Srivastava, R.A.K. Life-Style-Induced Metabolic Derangement and Epigenetic Changes Promote Diabetes and Oxidative Stress Leading to NASH and Atherosclerosis Severity. J. Diabetes Metab. Disord. 2018, 17, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Mohammed, S.A.; Ambrosini, S.; Paneni, F. Epigenetic Processing in Cardiometabolic Disease. Atherosclerosis 2019, 281, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Bungau, S.; Behl, T.; Tit, D.M.; Banica, F.; Bratu, O.G.; Diaconu, C.C.; Nistor-Cseppento, C.D.; Bustea, C.; Aron, R.A.C.; Vesa, C.M. Interactions between Leptin and Insulin Resistance in Patients with Prediabetes, with and without NAFLD. Exp. Ther. Med. 2020, 20, 197. [Google Scholar] [CrossRef]
- Gheorghe, G.; Bungau, S.; Ceobanu, G.; Ilie, M.; Bacalbasa, N.; Bratu, O.G.; Vesa, C.M.; Gaman, M.A.; Diaconu, C.C. The Non-Invasive Assessment of Hepatic Fibrosis. J. Formos. Med. Assoc. 2021, 120, 794–803. [Google Scholar] [CrossRef]
- Ampuero, J.; Gallego-Durán, R.; Romero-Gómez, M. Association of NAFLD with Subclinical Atherosclerosis and Coronary-Artery Disease: Meta-Analysis. J. Rev. Esp. Enferm. Dig. 2015, 107, 10–16. [Google Scholar]
- Yoosoo, C.; Seungho, R.; Ki-Chul, S.; Yong, K.C.; Eunju, S.; Han-Na, K.; Hyun-Suk, J.; Kyung, E.Y.; Jiin, A.; Hocheol, S.; et al. Alcoholic and Non-Alcoholic Fatty Liver Disease and Associations with Coronary Artery Calcification (CAC): Evidence from the Kangbuk Samsung Health Study. Gut 2019, 68, 1667–1675. [Google Scholar]
- Treeprasertsuk, S.; Lopez-Jimenez, F.; Lindor, K.D. Nonalcoholic Fatty Liver Disease and the Coronary Artery Disease. Dig. Sci. 2011, 56, 35–45. [Google Scholar] [CrossRef]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Järvinen Hannele, Y. Increased Liver Fat, Impaired Insulin Clearance, and Hepatic and Adipose Tissue Insulin Resistance in Type 2 Diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef]
- Tarantino, G.; Caputi, A. JNKs, Insulin Resistance and Inflammation: A Possible Link between NAFLD and Coronary Artery Disease. World J. Gastroenterol. 2011, 17, 3785–3794. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of b Cell Mass in Response to High-Fat Diet. Cell. Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef]
- Weickert, M.O. Signalling Mechanisms Linking Hepatic Glucose and Lipid Metabolism. Pfeiffer. AF Diabetol. 2006, 49, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and Tumor Necrosis Factor-Alpha Mediate Free Fatty Acid-Induced Insulin Resistance in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef]
- Yilmaz, Y.; Kurt, R.; Gurdal, A.; Alahdaba, Y.O.; Yonal, O.; Senates, E.; Polat, N.; Erend, F.; Imeryuz, N.; Oflaz, H. Circulating Vaspin Levels and Epicardial Adipose Tissue Thickness Are Associated with Impaired Coronary Flow Reserve in Patients with Nonalcoholic Fatty Liver Disease. Atherosclerosis 2011, 217, 125–129. [Google Scholar] [CrossRef]
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernandez-Guerra, M.; Mayorga, M.; Dominguez-Diez, A.; Fernandez -Escalante, J.C.; Pons-Romero, F. Gene Expression of Tumor Necrosis Factor and TNF-Receptors, P55 and P75, in Nonalcoholic Steatohepatitis Patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-Induced Insulin Resistance in Mice Lacking Adiponectin/ACRP30. Nat. Med. 2002, 8, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.L.; Cooper, G.J.S. The Fat-Derived Hormone Adiponectin Alleviates Alcoholic and Nonalcoholic Fatty Liver Diseases in Mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [PubMed]
- McKimmie, R.L.; Daniel, K.R.; Carr, J.J.; Bowden, D.W.; Freedman, B.I.; Register, T.C.; Hsu, F.C.; Lohman, K.K.; Weinberg, R.B.; Wagenknecht, L.E. Hepatic Steatosis and Subclinical Cardiovascular Disease in a Cohort Enriched for Type 2 Diabetes: The Diabetes Heart Study. Am. J. Gastroenterol. 2008, 103, 3029–3035. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, F.; Sugiyama, S.; Kojima, S.; Maruyoshi, H.; Funahashi, T.; Sakamoto, T.; Yoshimura, K.; Kimura, K.; Umemura, S.; Ogawa, H. Hypoadiponectinemia Is Associated with Impaired Glucose Tolerance and Coronary Artery Disease in Non-Diabetic Men. Circ. J. 2007, 71, 1703–1709. [Google Scholar] [CrossRef][Green Version]
- Edmison, J.; McCullough, A.J. Pathogenesis of Non-Alcoholic Steatohepatitis: Human Data. Clin. Liver Dis. 2007, 11, 75–104. [Google Scholar] [CrossRef]
- Solga, S.F.; Diehl, A.M. Nonalcoholic Fatty Liver Disease: Lumen-Liver Interactions and Possible Role for Probiotics. J. Hepatol. 2003, 38, 681–687. [Google Scholar] [CrossRef]
- Wanless, I.R.; Bargman, J.M.; Oreopoullos, D.G.; Vas, S.I. Subcapsular Steatonecrosis in Response to Peritoneal Insulin Deliver: A Clue to the Pathogenesis of Steatonecrosis in Obesity. Mod. Pathol. 1989, 2, 69–74. [Google Scholar]
- Khalili, K.; Lan, F.P.; Hanbidge, A.E.; Muradali, D.; Oreopoulos, D.G.; Wanless, I.R. Hepatic Subcapsular Steatosis in Response to Intraperitoneal Insulin Delivery: CT Findings and Prevalence. AJR Am. J. Roentgenol. 2003, 180, 1601–1604. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Kalyankar, M.; Wu, X. Insulin Action Is Facilitated by Insulin-Stimulated Reactive Oxygen Species with Multiple Potential Signaling Targets. Diabetes 2005, 54, 311–321. [Google Scholar] [CrossRef]
- Li, X.L.; Man, K.; Ng, K.T.; Lee, T.K.; Lo, C.M.; Fan, S.T. Insulin in UW Solution Exacerbates Hepatic Ischemia/Reperfusion Injury by Energy Depletion through the IRS-2/SREBP-1c Pathway. Liver Transpl. 2004, 10, 1172–1182. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Fargion, S.; Mattioli, M.; Fracanzani, A.L.; Sampietro, M.; Tavazzi, D.; Fociani, P.; Taioli, E.; Valenti, L.; Fiorelli, G. Hyperferritinemia, Iron Overload, and Multiple Metabolic Alterations Identify Patients at Risk for Nonalcoholic Steatohepatitis. Am. J. Gastroenterol. 2001, 96, 2448–2455. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Real, J.M.; Casamitjana-Abella, R.; Ricart-Engel, W.; Arroyo, E.; Balanca, R.; Casamitjana-Abella, R.; Cabrero, D.; Fernandez-Castaner, M.; Soler, J. Serum Ferritin as a Component of the Insulin Resistance Syndrome. Diabetes Care 1998, 21, 62–68. [Google Scholar] [CrossRef]
- Mendler, M.-H.; Turlin, B.; Moirand, R.; Jouanolle, A.-M.; Sapey, T.; Guyader, D.; le Gall, J.-Y.; Brissot, P.; David, V.; Deugnier, Y. Insulin Resistance-Associated Hepatic Iron Overload. Gastroenterology 1999, 117, 1155–1163. [Google Scholar] [CrossRef]
- Riquelme, A.; Soza, A.; Nazal, L.; Martinez, G.; Kolbach, M.; Patillo, A.; Arellano, M.; Duarte, I.; Martinez, J.; Molgo, M.; et al. Histological Resolution of Steatohepatitis after Iron Depletion. Dig. Sci. 2004, 49, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.D. Nonalcoholic Steatosis and Steatohepatitis. I. Molecular Mechanism for Polyunsaturated Fatty Acid Regulation of Gene Transcription. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G865–G869. [Google Scholar] [CrossRef]
- Tafani, M.; Schneider, T.G.; Pastorino, J.G.; Farber, J.L. Cytochrome Dependent Activation of Caspase 3 by Tumor Necrosis Factor Requires Induction of the Mitochondrial Permeability Transition. Am. J. Pathol. 2000, 156, 2111–2121. [Google Scholar] [CrossRef]
- Pastorini, J.G.; Simbula, G.; Yamamoto, K.; Glascott, P.A., Jr.; Rothman, R.J.; Farber, J.L. Cytotoxicity of TNF Depends on Induction of the Mitochondrial Permeability Transition. J. Biochem. 1996, 271, 29792–29798. [Google Scholar]
- Paradis, V.; Perle, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High Glucose and Hyperinsulinemia Stimulate Connective Tissue Growth Factor Expression: A Potential Mechanism Involved in Progression to Fibrosis in Nonalcoholic Steatohepatitis. Hepatology 2001, 74, 738–744. [Google Scholar] [CrossRef]
- Poli, G. Pathogenesis of Liver Fibrosis: The Role of Oxidative Stress. Mol Asp. Med. 2000, 21, 49–98. [Google Scholar] [CrossRef]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic Body Engulfment by a Human Stellate Cell Line Is Profibrogenic. Lab. Investig. 2003, 83, 655–663. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte Apoptosis and FAS Expression Are Prominent Features of Human Nonalcoholic Steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Li, Z.; Oben, J.A.; Yang, S.; Lin, H.; Stafford, E.A.; Soloski, M.J.; Thomas, S.A.; Diehl, A.M. Norepinephrine Regulates Hepatic Innate Immune System in Leptin Deficient Mice with Nonalcoholic Steatohepatitis. Hepatology 2004, 40, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Käräjämäki, A.J.; Olli-Pekka, P.; Markku, S.; Kesäniemi, Y.A.; Heikki, H.; Ukkola, O. Non-Alcoholic Fatty Liver Disease as a Predictor of Atrial Fibrillation in Middle-Aged Population (OPERA Study). PLoS ONE 2015, 10, e0142937. [Google Scholar] [CrossRef]
- Targher, G.; Valbuso, F.; Bonapace, S.; Bertolini, L.; Zenari, L.; Rodella, S.; Zoppini, G.; Mantovani, W.; Barbieri, E.; Byrne, C.D. Non-Alcoholic Fatty Liver Disease Is Associated with an Increased Incidence of Atrial Fibrillation in Patients with Type 2 Diabetes. Clin. Sci. 2013, 125, 301–309. [Google Scholar] [CrossRef]
- Hung, C.S.; Tseng, P.H.; Tu, C.H.; Chen, C.C.; Liao, W.C.; Lee, Y.C.; Chiu, H.M.; Lin, H.J.; Ho, Y.L.; Yang, W.S.; et al. Nonalcoholic Fatty Liver Disease Is Associated with QT Prolongation in the General Population. J. Am. Heart Assoc. 2015, 4, e001820. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Rigamonti, A.; Bonapace, S.; Bolzan, B.; Pernigo, M.; Morani, F.; Giovanni, L.; Bergamini, C.; Bertolini, L.; Valbusa, F.; et al. Nonalcoholic Fatty Liver Disease Is Associated with Ventricular Arrhythmias in Patients with Type 2 Diabetes Referred for Clinically Indicated 24-h Holter Monitoring. Diabetes Care 2016, 39, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Rigolon, R.; Bonapace, S.; Morani, G.; Zoppini, G.; Bonora, E.; Targher, G. Nonalcoholic Fatty Liver Disease Is Associated with an Increased Risk of Heart Block in Hospitalized Patients with Type 2 Diabetes Mellitus. PLoS ONE 2017, 12, e0185459. [Google Scholar] [CrossRef]
- Tsang, T.S.; Barnes, M.E.; Miyasaka, Y.; Cha, S.S.; Bailey, K.R.; Verzosa, G.C.; Seward, J.B.; Gersh, B.J. Obesity as a Risk Factor for the Progression of Paroxysmal to Permanent Atrial Fibrillation: A Longitudinal Cohort Study of 21 Years. Eur. Heart J. 2008, 29, 2227–2233. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. Diabetologia 2016, 59, 1141–1144. [Google Scholar] [CrossRef]
- Graner, M.; Nyman, K.; Siren, R.; Pentikainen, M.O.; Lundbom, J.; Hakkarainen, A.; Lauerma, K.; Lundbom, N.; Nieminen, M.S.; Taskinen, M.R. Ectopic Fat Depots and Left Ventricular Function in Nondiabetic Men with Nonalcoholic Fatty Liver Disease. Circ. Cardiovasc. Imaging 2014, 8, e001979. [Google Scholar] [CrossRef] [PubMed]
- Käräjämäki, A.J.; Hukkanen, J.; Ukkola, O. The Association of Non-Alcoholic Fatty Liver Disease and Atrial Fibrillation: A Review. Ann. Med. 2018, 50, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Hung, C.S.; Wu, Y.W.; Lee, Y.C.; Lin, Y.H.; Lin, C.; Lo, M.T.; Chan, C.C.; Ma, H.P.; Ho, Y.L.; et al. Influence of Non-Alcoholic Fatty Liver Disease on Autonomic Changes Evaluated by the Time Domain, Frequency Domain, and Symbolic Dynamics of Heart Rate Variability. PLoS ONE 2013, 8, e61803. [Google Scholar] [CrossRef]
- Park, H.W.; Shen, M.J.; Lin, S.F.; Fishbein, M.C.; Chen, L.S.; Chen, P.S. Neural Mechanisms of Atrial Fibrillation. Curr. Opin. Cardiol. 2012, 27, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lip, G.Y.; Apostolakis, S. Inflammation in Atrial Fibrillation. J. Am. Coll. Cardiol. 2012, 60, 2263–2270. [Google Scholar] [CrossRef]
- Ding, Y.H.; Ma, Y.; Qian, L.Y.; Xu, Q.; Wang, L.H.; Huang, D.S.; Zou, H. Linking Atrial Fibrillation with Non-Alcoholic Fatty Liver Disease: Potential Common Therapeutic Targets. Oncotarget 2017, 8, 60673–60683. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chen, Y.C.; Chen, J.H.; Chen, S.A.; Chen, Y.J. Adipocytes Modulate the Electrophysiology of Atrial Myocytes: Implications in Obesity-Induced Atrial Fibrillation. Basic Res. Cardiol. 2012, 107, 293. [Google Scholar] [CrossRef]
- Chung, M.K.; Martin, D.O.; Sprecher, D.; Wazni, O.; Kanderian, A.; Carnes, C.A.; Bauer, J.A.; Tchou, P.J.; Niebauer, M.J.; Natale, A.; et al. C-Reactive Protein Elevation in Patients with Atrial Arrhythmias: Inflammatory Mechanisms and Persistence of Atrial Fibrillation. Circulation 2001, 104, 2886–2891. [Google Scholar] [CrossRef]
- Mantovani, A.; Pernigo, M.; Bergamini, C.; Bonapace, S.; Lipari, P.; Valbusa, F.; Bertolini, L.; Zenari, L.; Pichiri, I.; Dauriz, M.; et al. Heart Valve Calcification in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Metabolism 2015, 64, 879–887. [Google Scholar] [CrossRef]
- Mantovani, A. Nonalcoholic Fatty Liver Disease (NAFLD) and Risk of Cardiac Arrhythmias: A New Aspect of the Liver-Heart Axis. J. Clin. Transl. Hepatol. 2017, 5, 134–141. [Google Scholar] [CrossRef]
- Ballestri, S.; Lonardo, A.; Bonapace, S.; Byrne, C.D.; Loria, P.; Targher, G. Risk of Cardiovascular, Cardiac and Arrhythmic Complications in Patients with Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 1724–1745. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G.; Lattuada, G.; De Cobelli, F.; Esposito, A.; Belloni, E.; Ntali, G.; Ragogna, F.; Canu, T.; Scifo, P.; Del Maschio, A.; et al. Increased Mediastinal Fat and Impaired Left Ventricular Energy Metabolism in Young Men with Newly Found Fatty Liver. Hepatology 2008, 47, 51–58. [Google Scholar] [CrossRef]
- Camici, P.; Ferrannini, E.; Opie, L.H. Myocardial Metabolism in Ischemic Heart Disease: Basic Principles and Application to Imaging by Positron Emission Tomography. Prog. Cardiovasc. Dis. 1989, 32, 217–238. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; McNulty, P.; Young, M.E. Adaptation and Maladaptation of the Heart in Diabetes: Part I: General Concepts. Circulation 2002, 105, 1727–1733. [Google Scholar] [CrossRef]
- Alp, H.; Karaarslan, S.; Eklioğlu, B.S.; Atabek, M.E.; Altın, H.; Baysal, T. Association between Nonalcoholic Fatty Liver Disease and Cardiovascular Risk in Obese Children and Adolescents. Can. J. Cardiol. 2013, 29, 1118–1125. [Google Scholar] [CrossRef]
- De Simone, G.; Richard, B.D.; Marcello, C.; Mary, J.R.; Elisa, T.L.; Helaine, E.R.; Barbara, V.H. Metabolic Syndrome and Left Ventricular Hypertrophy in the Prediction of Cardiovascular Events-The Strong Heart Study. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 98–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peterson, L.R.; Herrero, P.; Schechtman, K.B.; Racette, S.B.; Waggoner, A.D.; Kisrieva-Ware, Z.; Dence, C.; Klein, S.B.; Marsala, J.; Meyer, T.; et al. Effect of Obesity and Insulin Resistance on Myocardial Substrate Metabolism and Efficiency in Young Women. Circulation 2004, 109, 2191–2196. [Google Scholar] [CrossRef]
- Borges-Canha, M.; Neves, J.S.; Libâni, D.; Von-Hafe, M.; Vale, C.; Araújo-Martins, M.; Leite, A.R.; Pimentel-Nunes, P.; Davide, C.; Adelino, L.M. Association between Nonalcoholic Fatty Liver Disease and Cardiac Function and Structure—A Meta-Analysis. Endocrine 2019, 66, 467–476. [Google Scholar] [CrossRef]
- Kocabay, G.; Karabay, C.Y.; Colak, Y.; Oduncu, V.; Kalayci, A.; Akgun, T.; Guler, A.; Kirma, C. Left Atrial Deformation Parameters in Patients with Non-Alcoholic Fatty Liver Disease: A 2D Speckle Tracking Imaging Study. Clin. Sci. 2014, 126, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Mondillo, S.; Cameli, M.; Caputo, M.L.; Lisi, M.; Palmerini, E.; Padeletti, M.; Ballo, P. Early Detection of Left Atrial Strain Abnormalities by Speckle-Tracking in Hypertensive and Diabetic Patients with Normal Left Atrial Size. J. Am. Soc. Echocardiogr. 2011, 24, 898–908. [Google Scholar] [CrossRef]
- Bekler, A.; Gazi, E.; Erbag, G.; Binnetoglu, E.; Barutcu, A.; Sen, H.; Temiz, A.; Altun, B. Right Ventricular Function and Its Relationship with Grade of Hepatosteatosis in Non-Alcoholic Fatty Liver Disease. Cardiovasc. J. Afr. 2015, 26, 109–113. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Mantovani, A.; Tilg, H.; Targher, G. Risk of Cardiomyopathy and Cardiac Arrhythmias in Patients with Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Volzke, H.; Haring, R.; Lorbeer, R.; Wallaschofski, H.; Reffelmann, T.; Empen, K.; Rettig, R.; John, U.; Felix, S.B.; Dorr, M. Heart Valve Sclerosis Predicts All-Cause and Cardiovascular Mortality. Atherosclerosis 2010, 209, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the Early Lesion of ‘Degenerative’ Valvular Aortic Stenosis. Histological and Immunohistochemical Studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef]
- Goto, T.; Onuma, T.; Takebe, K.; Kral, J.G. The Influence of Fatty Liver on Insulin Clearance and Insulin Resistance in Non-Diabetic Japanese Subjects. Int. J. Obes. Relat. Metab. Disord. 1995, 19, 841–845. [Google Scholar]
- Paulista, M.; Marcello, R.; Baumeister, S.E.; Dörr, J.M.; Wallaschofski, H.; Völzke, H.; Lieb, W. Hepatic Steatosis Is Associated With Aortic Valve Sclerosis in the General Population The Study of Health in Pomerania (SHIP). Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1690–1695. [Google Scholar]
- Paulus, W.J.; Tschope, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin Signaling Coordinately Regulates Cardiac Size, Metabolism, and Contractile Protein Isoform Expression. J. Clin. Investig. 2002, 109, 629–639. [Google Scholar] [CrossRef]
- Dhingra, R.; Gona, P.; Wang, T.J.; Fox, C.S.; D’Agostino, R.B.; Vasan, R.S. Serum Gamma-Glutamyl Transferase and Risk of Heart Failure in the Community. Arter. Thromb. Vasc. Biol. 2010, 30, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Whincup, P.H.; Shaper, A.G.; Lennon, L.; Sattar, N. Γ-Glutamyltransferase, Hepatic Enzymes, and Risk of Incident Heart Failure in Older Men. Arter. Thromb. Vasc. Biol. 2012, 32, 830–835. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Emdin, M.; Pompella, A.; Paolicchi, A. γ-Glutamyltransferase, Atherosclerosis, and Cardiovascular Disease: Triggering Oxidative Stress within the Plaque. Circulation 2005, 112, 2078–2208. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nagata, C.; Takeda, J.; Sarui, H.; Kawahito, Y.; Yoshida, N.; Suetsugu, A.; Kato, T.; et al. Nonalcoholic Fatty Liver Disease Is a Novel Predictor of Cardiovascular Disease. World J. Gastroenterol. 2007, 13, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Abdeldyem, S.M.; Goda, T.; Khodeir, S.A.; Abou, S.S.; Abd-Elsalam, S. Nonalcoholic Fatty Liver Disease in Patients with Acute Ischemic Stroke Is Associated with More Severe Stroke and Worse Outcome. J. Clin. Lipidol. 2017, 11, 915–919. [Google Scholar] [CrossRef] [PubMed]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Cardio-Metabolic Disorders in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 2215. [Google Scholar] [CrossRef]
- Bots, M.L.; Salonen, J.; Elwood, P.; Nikitin, Y.; Freire, D.; Inzitari, D.; Sivenius, J.; Trichopoulou, A.; Tuomilehto, J.; Koudstaal, P.; et al. Gamma-Glutamyltransferase and Risk of Stroke: The EUROSTROKE Project. J. Epidemiol. Community Health 2002, 56, i25–i29. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, A.; Emdin, M.; Ghliozeni, E.; Ciancia, E.; Passino, C.; Popoff, G.; Pompella, A. Images in Cardiovascular Medicine, Human Atherosclerotic Plaques Contain Gamma-Glutamyl Transpeptidase Enzyme Activity. Circulation 2004, 109, 1440. [Google Scholar] [CrossRef]
- Emdin, M.; Passino, C.; Franzini, M.; Paolicchi, A.; Pompella, A. γ-Glutamyltransferase and Pathogenesis of Cardiovascular Diseases. Future Cardiol. 2007, 263–270. [Google Scholar] [CrossRef]
- Khalaf, M.R.; Hayhoe, F.G. Cytochemistry of Gamma-Glutamyltransferase in Haemic Cells and Malignancies. Histochem. J. 1987, 19, 385–395. [Google Scholar] [CrossRef]
- Stark, A.A.; Zeiger, E.; Pagano, D.A. Glutathione Metabolism by Gamma-Glutamyltranspeptidase Leads to Lipid Peroxidation: Characterization of the System and Relevance to Hepatocarcinogenesis. Carcinogenesis 1993, 14, 183–189. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.; John, F. Role of Oxidative Modifications in Atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Ciavarella, A.; Gnocchi, D.; Custodero, C.; Lenato, G.M.; Fiore, G.; Sabbà, C.; Mazzocca, A. Translational Insight into Prothrombotic State and Hypercoagulation in Nonalcoholic Fatty Liver Disease. Thromb. Res. 2021, 198, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.D.; Tufano, A.; Rusolillo, A.; Di Minno, G.; Tarantino, G. High Prevalence of Nonalcoholic Fatty Liver in Patients with Idiopathic Venous Thromboembolism. World J. Gastroenterol. 2010, 28, 6119–6122. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.; Jonathan, G.; Shah, N.L.; Argo, C.K.; Pelletier, S.J.; Caldwell, S.H.; Northup, P.G. Northup Increased Risk of Portal Vein Thrombosis in Patients With Cirrhosis Due to Nonalcoholic Steatohepatitis. Liver Transpl. 2015, 21, 1016–1021. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Stroes, E.S.; Meijers, J.C.; Buller, H. Hypercoagulability in the Metabolic Syndrome. Curr. Opin. Pharmacol. 2005, 5, 155–159. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, J.E.; Song, S.H.; Kim, H.K. Influence of Blood Lipids on Global Coagulation Test Results. Ann. Lab. Med. 2015, 35, 15–21. [Google Scholar] [CrossRef]
- Yun, J.W.; Cho, Y.K.; Park, J.H.; Kim, H.J.; Park, D.I.; Sohn, C.I.; Jeon, W.K.; Kim, B.I. Abnormal Glucose Tolerance in Young Male Patients with Nonalcoholic Fatty Liver Disease. Liver Int. 2009, 29, 525–529. [Google Scholar] [CrossRef]
- Undas, A.; Wiek, I.; Stepien, E.; Zmudka, K.; Tracz, W. Hyperglycemia Is Associated with Enhanced Thrombin Formation, Platelet Activation, and Fibrin Clot Resistance to Lysis in Patients with Acute Coronary Syndrome. Diabetes Care 2008, 31, 1590–1595. [Google Scholar] [CrossRef]
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet Dysfunction in Type 2 Diabetes. Diabetes Care 2001, 24, 1476–1485. [Google Scholar] [CrossRef]
- Ceriello, A. Coagulation Activation in Diabetes Mellitus: The Role of Hyperglycaemia and Therapeutic Prospects. Diabetologia 1993, 36, 1119–1125. [Google Scholar] [CrossRef]
- Mitropoulos, K.A.; Miller, G.J.; Watts, G.F.; Durrington, P.N. Lipolysis of Triglyceride-Rich Lipoproteins Activates Coagulant Factor XII: A Study in Familial Lipoprotein-Lipase Deficiency. Atherosclerosis 1992, 95, 119–125. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic Profiling Reveals a Contribution of Gut Microbiota to Fatty Liver Phenotype in Insulin-Resistant Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of Gut-Flora-Dependent Metabolite Trimethylamine-Noxide, Betaine and Choline with Non-Alcoholic Fatty Liver Disease in Adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Sookoian, S.; Castano, G.O.; Burgueno, A.L.; Rosselli, M.S.; Gianotti, T.F.; Mallardi, P.; Martino, J.S.; Pirola, C.J. Circulating Levels and Hepatic Expression of Molecular Mediators of Atherosclerosis in Nonalcoholic Fatty Liver Disease. Atherosclerosis 2010, 209, 585–591. [Google Scholar] [CrossRef]
- Esmon, C.T. The Interactions between Inflammation and Coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Dole, V.S.; Bergmeier, W.; Patten, I.S.; Hirahashi, J.; Mayadas, T.N.; Wagner, D.D. PSGL-1 Regulates Platelet P-Selectin-Mediated Endothelial Activation and Shedding of P-Selectin from Activated Platelets. Thromb. Haemost. 2007, 98, 806–812. [Google Scholar] [CrossRef]
- Patil, R.; Sood, G.K. Non-Alcoholic Fatty Liver Disease and Cardiovascular Risk. World J. Gastrointest. Pathophysiol. 2017, 8, 51–58. [Google Scholar] [CrossRef]
- Abed, H.S.; Wittert, G.A.; Leong, D.P.; Shirazi, M.G.; Bahrami, B.; Middeldorp, M.E.; Lorimer, M.F.; Lau, D.H.; Antic, N.A.; Brooks, A.G.; et al. Effect of Weight Reduction and Cardiometabolic Risk Factor Management on Symptom Burden and Severity in Patients with Atrial Fibrillation: A Randomized Clinical Trial. JAMA 2013, 310, 2050–2060. [Google Scholar] [CrossRef]
- Averna, M. The Effect of Ezetimibe on NAFLD. Atheroscler. Suppl. 2015, 17, 27–34. [Google Scholar] [CrossRef]
- Abe, M.; Matsuda, M.; Kobayashi, H.; Miyata, Y.; Nakayama, Y.; Komuro, R.; Fukuhara, A.; Shimomura, I. Effects of Statins on Adipose Tissue Inflammation: Their Inhibitory Effect on MyD88-Independent IRF3/IFN-Beta Pathway in Macrophages. Arter. Thromb. Vasc. Biol. 2008, 28, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Parisi, V.; Petraglia, L.; D’Esposito, V.; Cabaro, S.; Rengo, G.; Caruso, A.; Grimaldi, M.G.; Baldascino, F.; De Bellis, A.; Vitale, D.; et al. Therapy Modulates Thickness and Inflammatory Profile of Human Epicardial Adipose Tissue. Int. J. Cardiol. 2019, 274, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.D.; Tang, B.P.; Guo, F.; Li, J.X.; Han, W.; Tang, Q.; Zhang, Y.Y. Effect of Atorvastatin on Left Atrial Function of Patients with Paroxysmal Atrial Fibrillation. Genet. Mol. Res. 2013, 12, 3488–3494. [Google Scholar] [CrossRef]
- Fauchier, L.; Pierre, B.; de Labriolle, A.; Grimard, C.; Zannad, N.; Babuty, D. Antiarrhythmic Effect of Statin Therapy and Atrial Fibrillation a Meta-Analysis of Randomized Controlled Trials. J. Am. Coll. Cardiol. 2008, 51, 828–835. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.; An, J.; Song, Y.; Lee, C.K.; Kim, K.; Kong, H. Alterations in Gut Microbiota by Statin Therapy and Possible Intermediate Effects on Hyperglycemia and Hyperlipidemia. Front. Microbiol. 2019, 10, 10. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D.; Schwenke, D.C.; Banerji, M.A.; Bray, G.A.; Buchanan, T.A.; Clement, S.C.; Henry, R.R.; Hodis, H.N.; Kitabchi, A.E.; et al. Pioglitazone for Diabetes Prevention in Ipaired Glucose Tolerance. N. Engl. J. Med. 2011, 364, 1104–1115. [Google Scholar] [CrossRef]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.A.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefebvre, P.J.; Murray, G.D.; et al. Secondary Prevention of MacroVascular Events in Patients with Type 2 Diabetes in the Proactive Study (Prospective PioglitAzone Clinical Trial in MacroVascular Events): A Randomised Controlled Trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-Analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Yu, R.; Zhang, L.P.; Wen, S.Y.; Wang, S.J.; Zhang, X.Y.; Xu, Q.; Kong, L.D. Dietary Fructose-Induced Gut Dysbiosis Promotes Mouse Hippocampal Neuroinflammation: A Benefit of Short-Chain Fatty Acids. Microbiome 2019, 7, 98. [Google Scholar] [CrossRef]
- Stumvoll, M.; Nurjhan, N.; Perriello, G.; Dailey, G.; Gerich, J.E. Metabolic Effects of Metformin in Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1995, 333, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Lomba, R.; Lutchman, G.; Kleiner, D.E.; Ricks, M.; Feld, J.J.; Borg, B.B.; Modi, A.; Nagabhyru, P.; Sumner, A.E.; Liang, T.J.; et al. Clinical Trial: Pilot Study of Metformin for the Treatment of Non-Alcoholic Steatohepatitis. Aliment. Pharmacol. Ther. 2009, 29, 172–182. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and Insulin Suppress Hepatic Gluconeogenesis through Phosphorylation of CREB Binding Protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef]
- Kohjima, M.; Higuchi, N.; Kato, M.; Koroh, K.; Yoshimoto, T.; Fujino, T.; Yada, R.; Yada, R.; Harada, N.; Enjoji, M. SREBP-1c, Regulated by the Insulin and AMPK Signaling Pathways, Plays a Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef]
- Nair, S.; Diehl, A.M.; Wiseman, M.; Farr, G.H.; Perrillo, R.P., Jr. Metformin in the Treatment of Non-Alcoholic Steatohepatitis: A Pilot Open Label Trial. Aliment. Pharmacol. Ther. 2004, 20, 23–28. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gentilcore, E.; Manini, R.; Natale, S.; Vanni, E.; Vilanova, N.; David, E.; Rizzetto, M.; Marchesini, G. A Randomized Controlled Trial of Metformin versus Vitamin E or Prescriptive Diet in Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2005, 100, 1082–1090. [Google Scholar] [CrossRef]
- De Oliveira, C.P.M.S.; Stefano, J.T.; De Siqueira, E.R.F.; Soares Silva, L.; Ferraz de Campos Marzo, D.; Lima, V.M.R.; Furuya, C.K.; Mello, E.S.; Souza, F.G.; Rabello, F.; et al. Combination of N-Acetylcysteine and Metformin Improves Histological Steatosis and Fibrosis in Patients with Non-Alcoholic Steatohepatitis. Hepatol. Res. 2008, 38, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Idilman, R.; Mizrak, D.; Corapcioglu, D.; Bektas, M.; Doganay, B.; Sayki, M.; Coban, S.; Erden, E.; Soykan, I.; Emral, R.; et al. Clinical Trial: Insulin-Sensitizing Agents May Reduce Consequences of Insulin Resistance in Individuals with Non-Alcoholic Steatohepatitis. Aliment. Pharmacol. Ther. 2008, 28, 200–208. [Google Scholar] [CrossRef]
- Janiec, D.J.; Jacobson, E.R.; Freeth, A.; Spaulding, L.; Blaszyk, H. Histologic Variation of Grade and Stage of Non-Alcoholic Fatty Liver Disease in Liver Biopsies. Obes. Surg. 2005, 15, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Haukeland, J.W.; Konopski, Z.; Eggesb, H.B.; von Volkmann, H.L.; Raschpichler, G.; Bjoro, K.; Haaland, T.; Loberg, E.M.; Birkeland, K. Metformin in Patients with Non-Alcoholic Fatty Liver Disease: A Randomized, Controlled Trial. Scand. J. Gastroenterol. 2009, 44, 853–860. [Google Scholar] [CrossRef]
- Omer, Z.; Cetinkalp, S.; Akyildiz, M.; Yilmaz, F.; Batur, Y.; Yilmaz, C.; Akarca, U. Efficacy of Insulin-Sensitizing Agents in Nonalcoholic Fatty Liver Disease. Eur. J. Gastroenterol. Hepatol. 2010, 22, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Nar, A.; Gedik, O. The Effect of Metformin on Leptin in Obese Patients with Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease. Acta Diabetol. 2009, 46, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Lonardo, A.; Bellentani, S.; Day, C.P.; Marchesini, G.; Carulli, N. Non-Alcoholic Fatty Liver Disease (NAFLD) and Cardiovascular Disease: An Open Question. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Kirpichnikov, D.; McFarlane, S.I.; Sowers, J.R. Metformin: An Update. Ann. Intern. Med. 2002, 137, 25–33. [Google Scholar] [CrossRef]
- Morrow, V.A.; Foufelle, F.; Connell, J.M.C.; Petrie, J.R.; Gould, G.W.; Salt, I.P. Direct Activation of AMP-Activated Protein Kinase Stimulates Nitric-Oxide Synthesis in Human Aortic Endothelial Cells. J. Biol. Chem. 2003, 278, 31629–31639. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Scorletti, E.; Mantzoros, C.S.; Targher, G. Efficacy and Safety of Anti-Hyperglycaemic Drugs in Patients with Non-Alcoholic Fatty Liver Disease with or without Diabetes: An Updated Systematic Review of Randomized Controlled Trials. Diabetes Metab. 2020, 46, 427–441. [Google Scholar] [CrossRef]
- Zhu, J.; Yu, X.; Zheng, Y.; Li, J.; Wang, Y.; Lin, Y.; He, Z.; Zhao, W.; Chen, C.; Qiu, K.; et al. Association of Glucose-Lowering Medications with Cardiovascular Outcomes: An Umbrella Review and Evidence MAP. Lancet Diabetes Endocrinol. 2020, 8, 192–205. [Google Scholar] [CrossRef]
- Nakamura, H.; Niwano, S.; Niwano, H.; Fukaya, H.; Murakami, M.; Kishihara, J.; Satoh, A.; Yoshizawa, T.; Ishizue, N.; Igarashi, T.; et al. Liraglutide Suppresses Atrial Electrophysiological Changes. Heart Vessel. 2019, 34, 1389–1393. [Google Scholar] [CrossRef]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte Lipolysis and Insulin Resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hazlehurst, J.M.; Hull, D.; Guo, K.; Borrows, S.; Yu, J.; Gough, S.C.; Newsome, P.N.; Tomlinson, J.W. Abdominal Subcutaneous Adipose Tissue Insulin Resistance and Lipolysis in Patients with Non-alcoholic Steatohepatitis. Diabetes Obes. Metab. 2014, 16, 651–660. [Google Scholar] [CrossRef]
- Sindhu, S.; Thomas, R.; Shihab, P.; Sriraman, D.; Behbehani, K.; Ahmad, R. Obesity Is a Positive Modulator of IL-6R and IL-6 Expression in the Subcutaneous Adipose Tissue: Significance for Metabolic Inflammation. PLoS ONE 2015, 10, e0133494. [Google Scholar] [CrossRef]
- Cnop, M.; Havel, P.J.; Utzschneider, K.M.; Carr, D.B.; Sinha, M.K.; Boyko, E.J.; Retzlaff, B.M.; Knopp, R.H.; Brunzell, J.D.; Kahn, S.E. Relationship of Adiponectin to Body Fat Distribution, Insulin Sensitivity and Plasma Lipoproteins: Evidence for Independent Roles of Age and Sex. Diabetologia 2003, 46, 459–469. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin Stimulates Glucose Utilization and Fatty-acid Oxidation by Activating AMP-activated Protein Kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond Insulin Resistance in NASH: TNF-alpha or Adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The Role of Hepatic Lipids in Hepatic Insulin Resistance and Type 2 Diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Jin, T. The Incretin Hormone GLP-1 and Mechanisms Underlying Its Secretion. J. Diabetes 2016, 8, 753–765. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Sathyapalan, T.; Sahebkar, A. Molecular Mechanisms by Which GLP-1 RA and DPP-4i Induce Insulin Sensitivity. Life Sci. 2019, 234, 116776. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D. Glucagon-like Peptide-1 Receptor Activation Stimulates Hepatic Lipid Oxidation and Restores Hepatic Signalling Alteration Induced by a High-fat Diet in Nonalcoholic Steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic Fatty Liver Disease and Diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef]
- Klonoff DC, B.J.; Nielsen, L.L.; Guan, X.; Bowlus, C.L.; Holcombe, J.H.; Wintle, M.E.; Maggs, D.G. Metabolic Effects of Two Years of Exenatide Treatment on Diabetes, Obesity, and Hepatic Biomarkers in Patients with Type 2 Diabetes: An Interim Analysis of Data from the Open-label, Uncontrolled Extension of Three Double-blind, Placebo-controlled Trials. Clin. Ther. 2007, 2, 139–153. [Google Scholar]
- Klonoff, D.C.; Buse, J.B.; Nielsen, L.L.; Guan, X.; Bowlus, C.L.; Holcombe, J.H.; Wintle, M.E.; Maggs, D.G. Exenatide Effects on Diabetes, Obesity, Cardiovascular Risk Factors and Hepatic Biomarkers in Patients with Type 2 Diabetes Treated for at Least 3 Years. Curr. Med. Res. Opin. 2008, 24, 275–286. [Google Scholar] [CrossRef]
- Castera, L.; Vilgrain, V.; Angulo, P. Noninvasive Evaluation of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Vuppalanchi, R.; Siddiqui, M.S.; Van Natta, M.L.; Hallinan, E.; Brandman, D.; Kowdley, K.; Neuschwander-Tetri, B.A.; Loomba, R.; Dasarathy, S.; Abdelmalek, M.; et al. Performance Characteristics of Vibration-controlled Transient Elastography for Evaluation of Nonalcoholic Fatty Liver Disease. Hepatology 2018, 67, 134–144. [Google Scholar] [CrossRef]
- Dong, Y.; Lv, Q.; Li, S.; Wu, Y.; Li, L.; Zhang, F.; Sun, X.; Tong, N. Efficacy and Safety of Glucagon-like Peptide-1 Receptor Agonists in Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 284–295. [Google Scholar] [CrossRef]
- Lv, X.; Dong, Y.; Hu, L.; Lu, F.; Zhou, C.; Qin, S. Glucagon-like Peptide-1 Receptor Agonists (GLP-1 RAs) for the Management of Nonalcoholic Fatty Liver Disease (NAFLD): A Systematic Review. Endocrinol. Diab. Metab. 2020, 3, e00163. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Fitchett, D.; Hantel, S.; George, J.T.; Zinman, B. Empagliflozin Is Associated with Improvements in Liver Enzymes Potentially Consistent with Reductions in Liver Fat: Results from Randomised Trials Including the EMPA-REG OUTCOME® Trial. Diabetologia 2018, 61, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Katsuno, K.; Isaji, M.; Nagasawa, T.; Buehrer, B.; Walker, S.; Wilkison, W.O.; Cheatham, B. Remogliflozin Etabonate Improves Fatty Liver Disease in Diet-Induced Obese Male Mice. J. Clin. Exp. Hepatol. 2015, 5, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Komiya, C.; Tsuchiya, K.; Shiba, K.; Miyachi, Y.; Furuke, S.; Shimazu, N.; Yamaguchi, S.; Kanno, K.; Ogawa, Y. Ipragliflozin Improves Hepatic Steatosis in Obese Mice and Liver Dysfunction in Type 2 Diabetic Patients Irrespective of Body Weight Reduction. PLoS ONE 2016, 11, e0151511. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, T.; Fushimi, N.; Kawai, M.; Yoshida, Y.; Hachiya, H.; Ito, S.; Kawai, H.; Ohashi, N.; Mori, A. Luseogliflozin Improves Liver Fat Deposition Compared to Metformin in Type 2 Diabetes Patients with Non-Alcoholic Fatty Liver Disease: A Prospective Randomized Controlled Pilot Study. Diabetes Obes. Metab. 2018, 20, 438–442. [Google Scholar] [CrossRef]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 Inhibitor), Alone or in Combination with Linagliptin (a DPP-4 Inhibitor), Prevents Steatohepatitis in a Novel Mouse Model of Non-Alcoholic Steatohepatitis and Diabetes. Diabetol. Metab. Syndr. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Sumida, Y.; Tanaka, S.; Mori, K.; Taketani, H.; Ishiba, H.; Hara, T.; Okajima, A.; Umemura, A.; Nishikawa, T.; et al. Effect of Sodium Glucose Cotransporter 2 Inhibitor on Liver Function Tests in Japanese Patients with Non-Alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. Hepatol. Res. 2017, 47, 1072–1078. [Google Scholar] [CrossRef]
- Sumida, Y.; Murotani, K.; Saito, M.; Tamasawa, A.; Osonoi, Y.; Yoneda, M.; Osonoi, T. Effect of Luseogliflozin on Hepatic Fat Content in Type 2 Diabetes Patients with Non-Alcoholic Fatty Liver Disease: A Prospective, Single-Arm Trial (LEAD Trial). Hepatol. Res. 2019, 49, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.A.; Johansson, L.; Kvarnström, M.; Moris, L.; Miliotis, T.; Forsberg, G.B.; Risérus, U.; Lind, L.; et al. Effects of Dapagliflozin and N-3 Carboxylic Acids on Non-Alcoholic Fatty Liver Disease in People with Type 2 Diabetes: A Double-Blind Randomised Placebo-Controlled Study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla Bertot, L.; Adams, L.A. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 20, 774. [Google Scholar] [CrossRef]
- Shah, A.G.; Lydecker, A.; Murray, K.; Tetri, B.N.; Contos, M.J.; Sanyal, A.J. NASH Clinical Research Network. Comparison of Noninvasive Markers of Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2009, 7, 1104–1112. [Google Scholar] [CrossRef]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD Fibrosis Score: A Noninvasive System That Identifies Liver Fibrosis in Patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef]
- Tobita, H.; Sato, S.; Miyake, T.; Ishihara, S.; Kinoshita, Y. Effects of Dapagliflozin on Body Composition and Liver Tests in Patients with Nonalcoholic Steatohepatitis Associated with Type 2 Diabetes Mellitus: A Prospective, Open-Label, Uncontrolled Study. Curr. Ther. Res. Clin. Exp. 2017, 87, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of Current Treatments on Liver Disease, Glucose Metabolism and Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease (NAFLD): A Systematic Review and Meta-Analysis of Randomized Trials. Diabetologia 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A Glucose-Responsive Transcription Factor That Regulates Carbohydrate Metabolism in the Liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef]
- Shimizu, M.; Suzuki, K.; Kato, K.; Jojima, T.; Iijima, T.; Murohisa, T.; Iijima, M.; Takekawa, H.; Usui, I.; Hiraishi, H.; et al. Evaluation of the Effects of Dapagliflozin, a Sodium-Glucose Co-Transporter-2 Inhibitor, on Hepatic Steatosis and Fibrosis Using Transient Elastography in Patients with Type 2 Diabetes and Non-Alcoholic Fatty Liver Disease. Diabetes Obes. Metab. 2019, 21, 285–292. [Google Scholar] [CrossRef]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis Prevents Diet-Induced Fatty Liver Injury and Hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef]
- Akuta, N.; Kawamura, Y.; Fujiyama, S.; Sezaki, H.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; Saitoh, S.; Suzuki, F.; Suzuki, Y.; et al. SGLT2 Inhibitor Treatment Outcome in Nonalcoholic Fatty Liver Disease Complicated with Diabetes Mellitus: The Long-Term Effects on Clinical Features and Liver Histopathology. Intern. Med. 2020, 59, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Oikonomoua, D.; Georgiopoulosb, G.; Katsib, V.; Kourekb, C.; Tsioufisb, C.; Alexopouloub, A.; Koutlic, E.; Tousoulisb, D. Non-Alcoholic Fatty Liver Disease and Hypertension: Coprevalent or Correlated? Eur. J. Gastroenterol. Hepatol. 2018, 30, 979–985. [Google Scholar] [CrossRef]
- Kaji, K.; Yoshiji, H.; Kitade, M.; Ikenaka, Y.; Noguchi, R.; Shirai, Y.; Aihara, Y.; Namisaki, T.; Yoshii, J.; Yanase, K.; et al. Combination Treatment of Angiotensin II Type I Receptor Blocker and New Oral Iron Chelator Attenuates Progression of Nonalcoholic Steatohepatitis in Rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 1094–1104. [Google Scholar] [CrossRef]
- Georgescu, E.F.; Ionescu, R.; Niculescu, M.; Mogoanta, L.; Vancica, L. Angiotensin-Receptor Blockers as Therapy for Mild-to-Moderate Hypertension-Associated Non-Alcoholic Steatohepatitis. World J. Gastroenterol. 2009, 15, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Tomita, K.; Kawai, T.; Yokoyama, H.; Shimada, A.; Kikuchi, M.; Hirose, H.; Ebinuma, H.; Irie, J.; Ojiro, K.; et al. Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY). Int. J. Endocrinol. 2013, 2013, 587140. [Google Scholar] [CrossRef]
- Randomized Global Phase 3 Study to Evaluate the Impact on NASH with Fibrosis of Obeticholic Acid Treatment (REGENERATE). U.S. National Library of Medicine. Clinical Trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02548351 (accessed on 31 January 2021).
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated Fibroblast Growth Factor 21 Analogue, in Patients with Non-Alcoholic Steatohepatitis: A Randomised, Double-Blind, Placebo-Controlled, Phase 2a Trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A Randomized, Placebo-Controlled Trial of Cenicriviroc for Treatment of Nonalcoholic Steatohepatitis with Fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jichitu, A.; Bungau, S.; Stanescu, A.M.A.; Vesa, C.M.; Toma, M.M.; Bustea, C.; Iurciuc, S.; Rus, M.; Bacalbasa, N.; Diaconu, C.C. Non-Alcoholic Fatty Liver Disease and Cardiovascular Comorbidities: Pathophysiological Links, Diagnosis, and Therapeutic Management. Diagnostics 2021, 11, 689. https://doi.org/10.3390/diagnostics11040689
Jichitu A, Bungau S, Stanescu AMA, Vesa CM, Toma MM, Bustea C, Iurciuc S, Rus M, Bacalbasa N, Diaconu CC. Non-Alcoholic Fatty Liver Disease and Cardiovascular Comorbidities: Pathophysiological Links, Diagnosis, and Therapeutic Management. Diagnostics. 2021; 11(4):689. https://doi.org/10.3390/diagnostics11040689
Chicago/Turabian StyleJichitu, Alexandra, Simona Bungau, Ana Maria Alexandra Stanescu, Cosmin Mihai Vesa, Mirela Marioara Toma, Cristiana Bustea, Stela Iurciuc, Marius Rus, Nicolae Bacalbasa, and Camelia Cristina Diaconu. 2021. "Non-Alcoholic Fatty Liver Disease and Cardiovascular Comorbidities: Pathophysiological Links, Diagnosis, and Therapeutic Management" Diagnostics 11, no. 4: 689. https://doi.org/10.3390/diagnostics11040689
APA StyleJichitu, A., Bungau, S., Stanescu, A. M. A., Vesa, C. M., Toma, M. M., Bustea, C., Iurciuc, S., Rus, M., Bacalbasa, N., & Diaconu, C. C. (2021). Non-Alcoholic Fatty Liver Disease and Cardiovascular Comorbidities: Pathophysiological Links, Diagnosis, and Therapeutic Management. Diagnostics, 11(4), 689. https://doi.org/10.3390/diagnostics11040689