
Diagnostic Immunohistochemistry of Soft Tissue and Bone Tumors: An Update on Biomarkers That Correlate with Molecular Alterations

Abstract
1. Introduction
2. Surrogate Markers of Gene Fusions
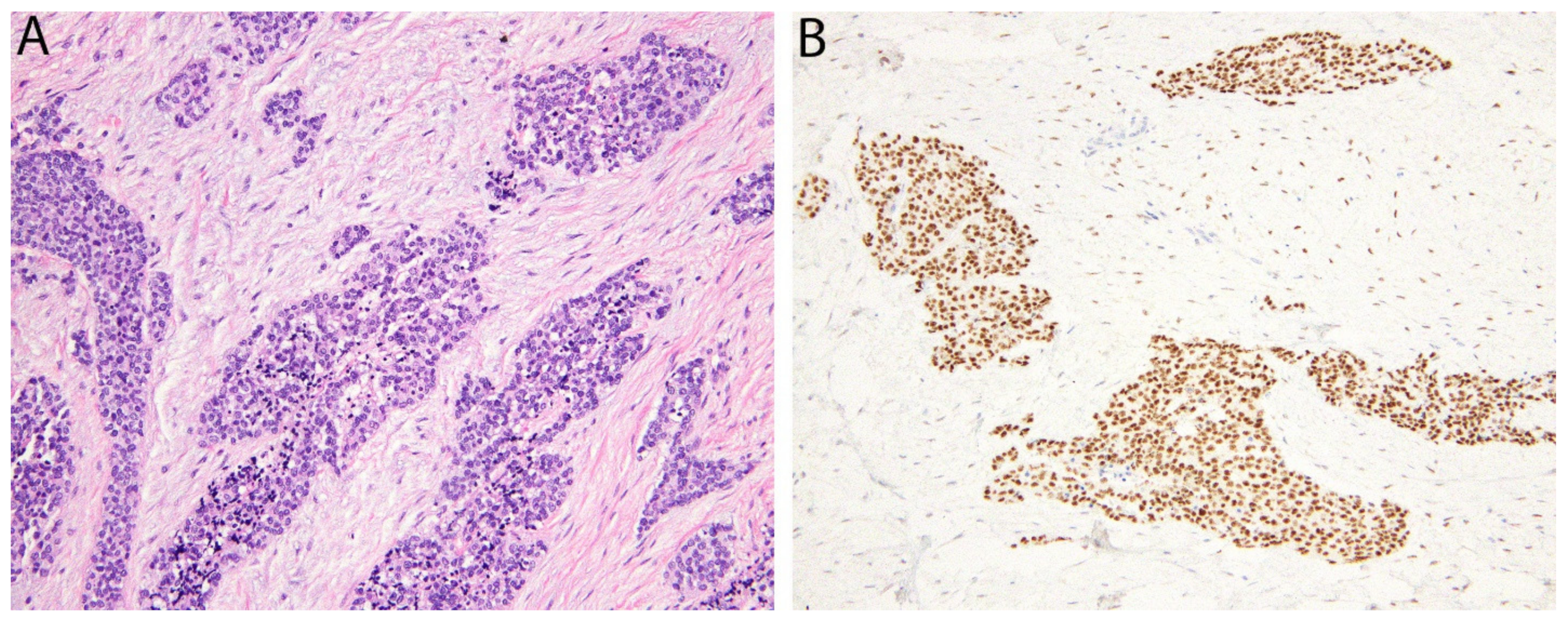
2.1. CCNB3 in BCOR-Rearranged Sarcoma
2.2. BCOR in BCOR-Rearranged Sarcoma
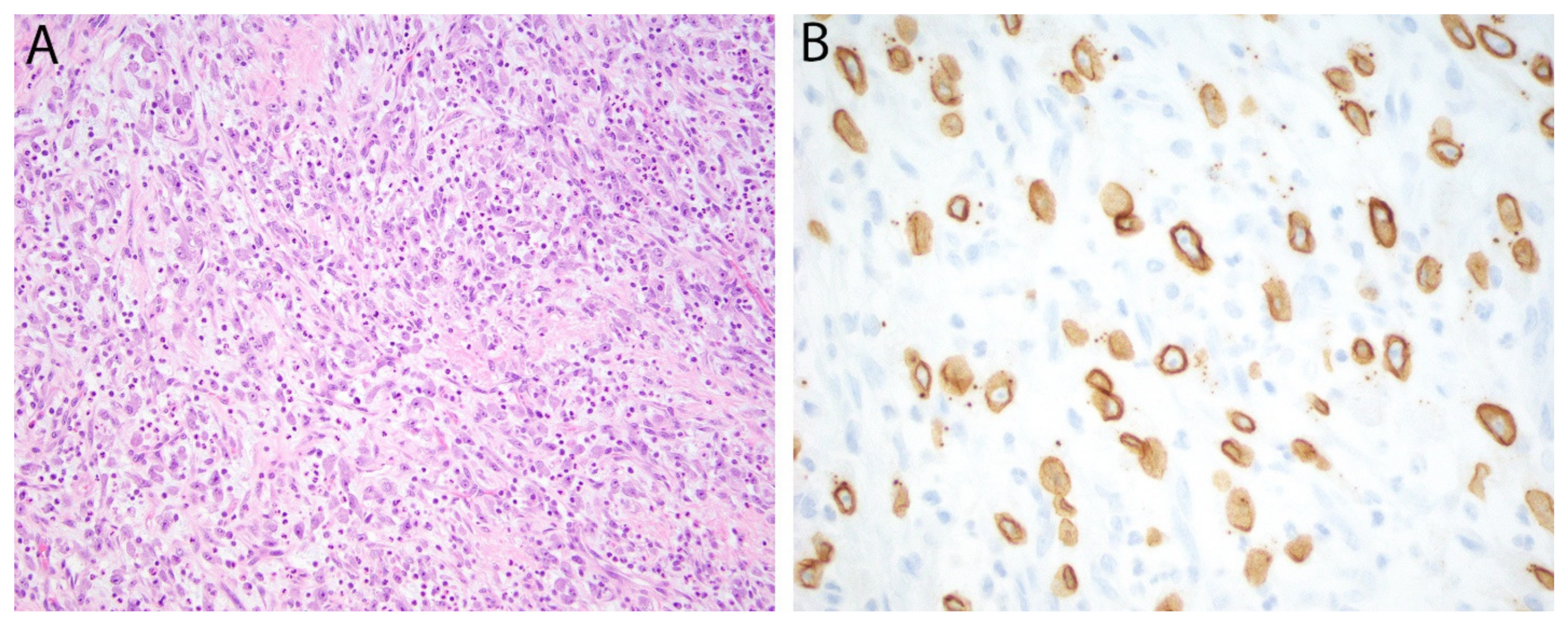
2.3. WT1 (C-Terminus) in Desmoplastic Small Round Cell Tumor
2.4. PAX3/7-FOXO1 in Alveolar Rhabdomyosarcoma
2.5. PAX3 in Biphenotypic Sinonasal Sarcoma
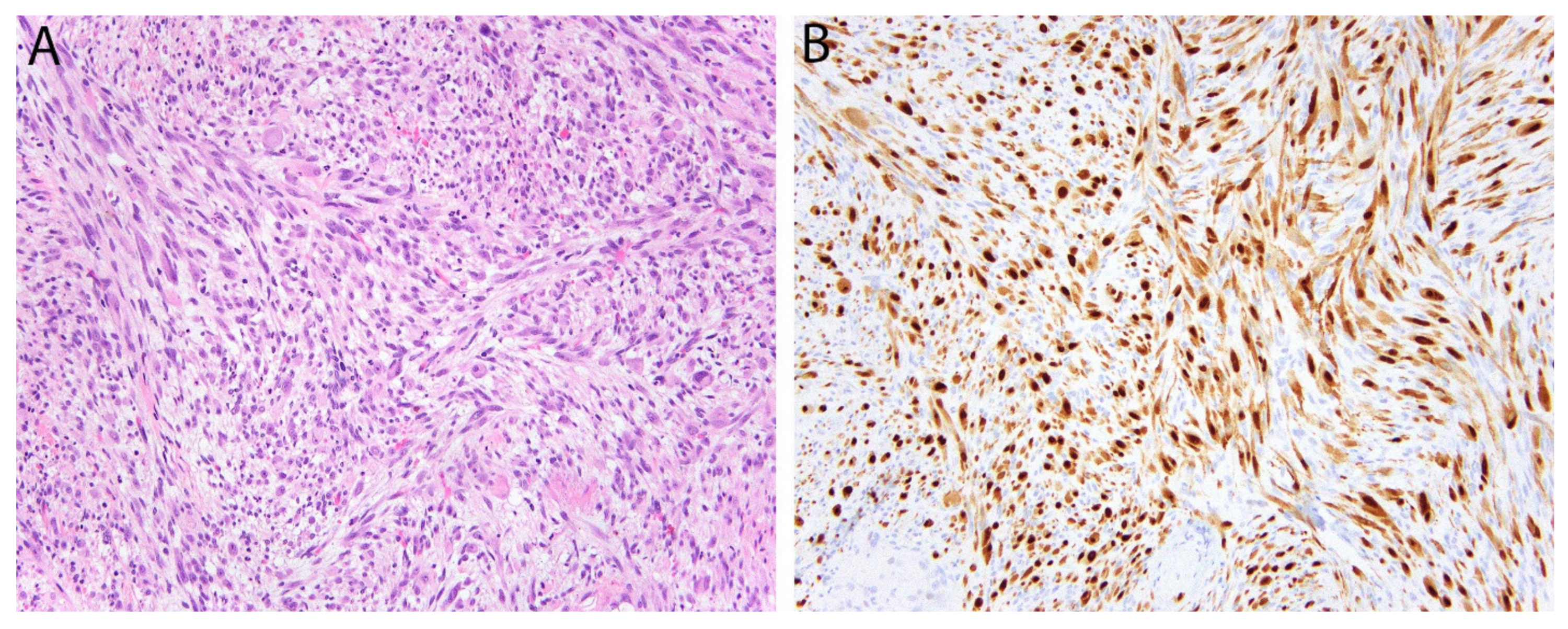
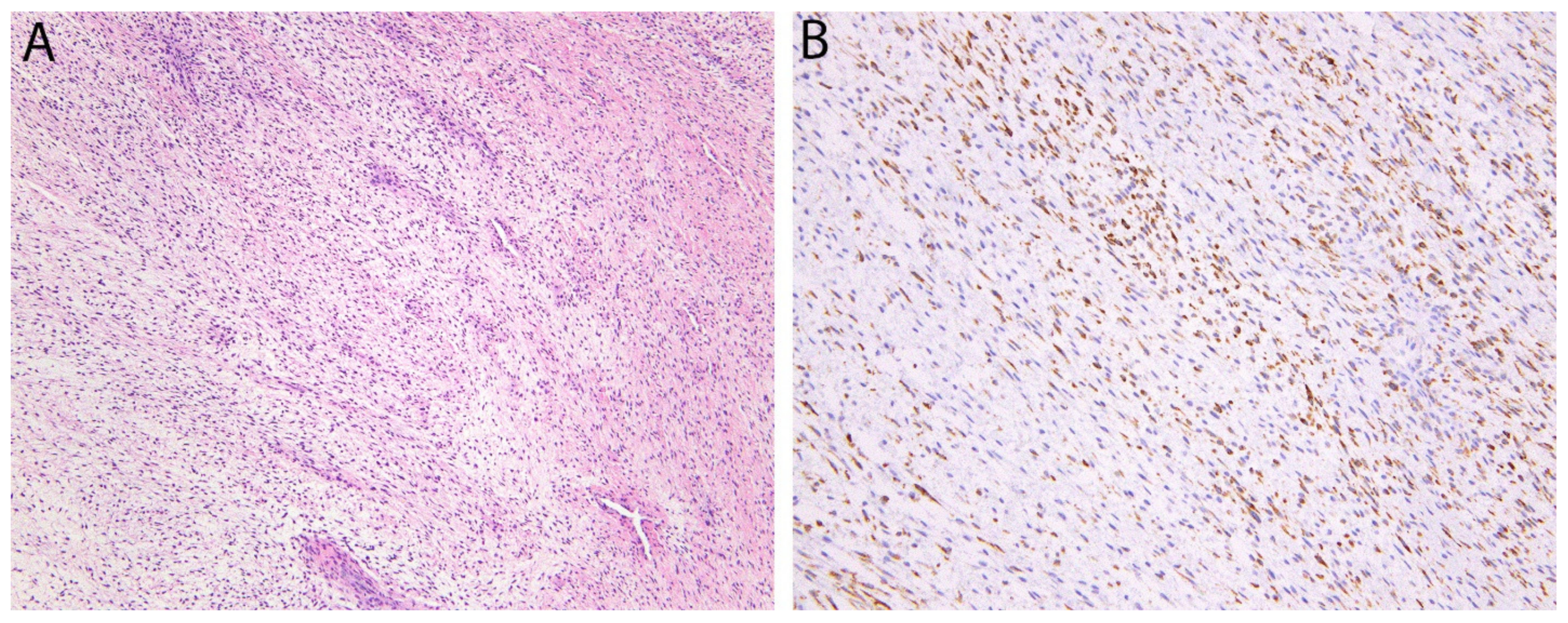
2.6. STAT6 in Solitary Fibrous Tumor
2.7. ALK in Inflammatory Myofibroblastic Tumor
2.8. Pan-Trk in Infantile Fibrosarcoma and NTRK-Rearranged Spindle Cell Neoplasms
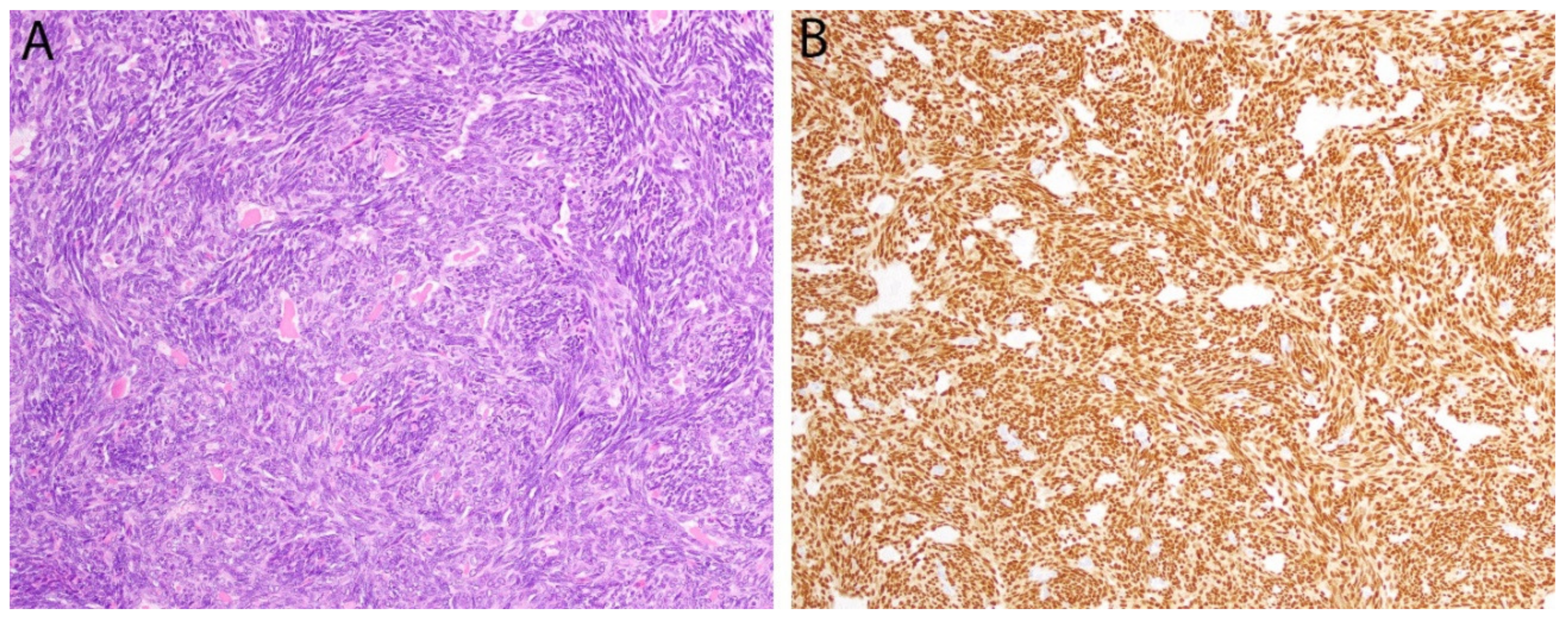
2.9. SS18-SSX/SSX C-Terminus in Synovial Sarcoma
2.10. CAMTA1 in Epithelioid Hemangioendothelioma
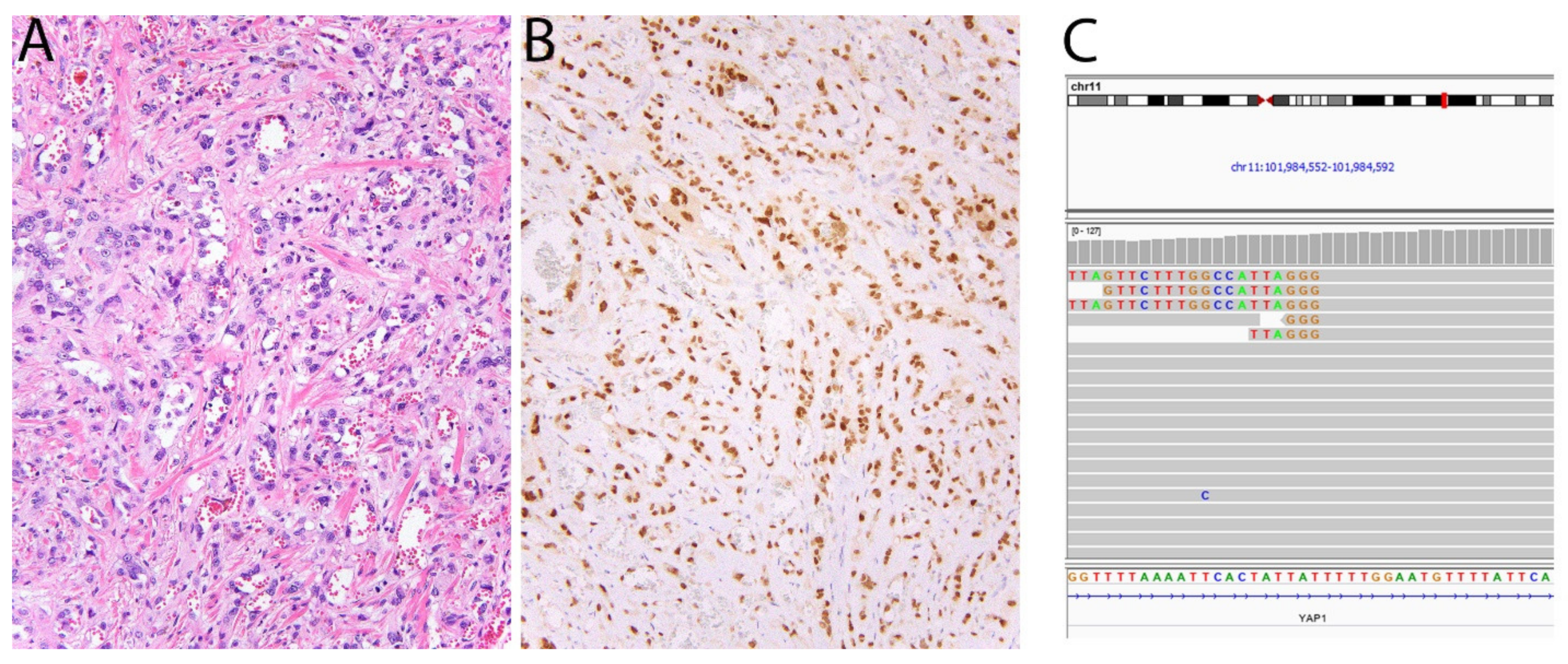
2.11. TFE3 in Alveolar Soft Part Sarcoma, PEComa, and Epithelioid Hemangioendothelioma
2.12. FOSB/FOS in Pseudomyogenic Hemangioendothelioma
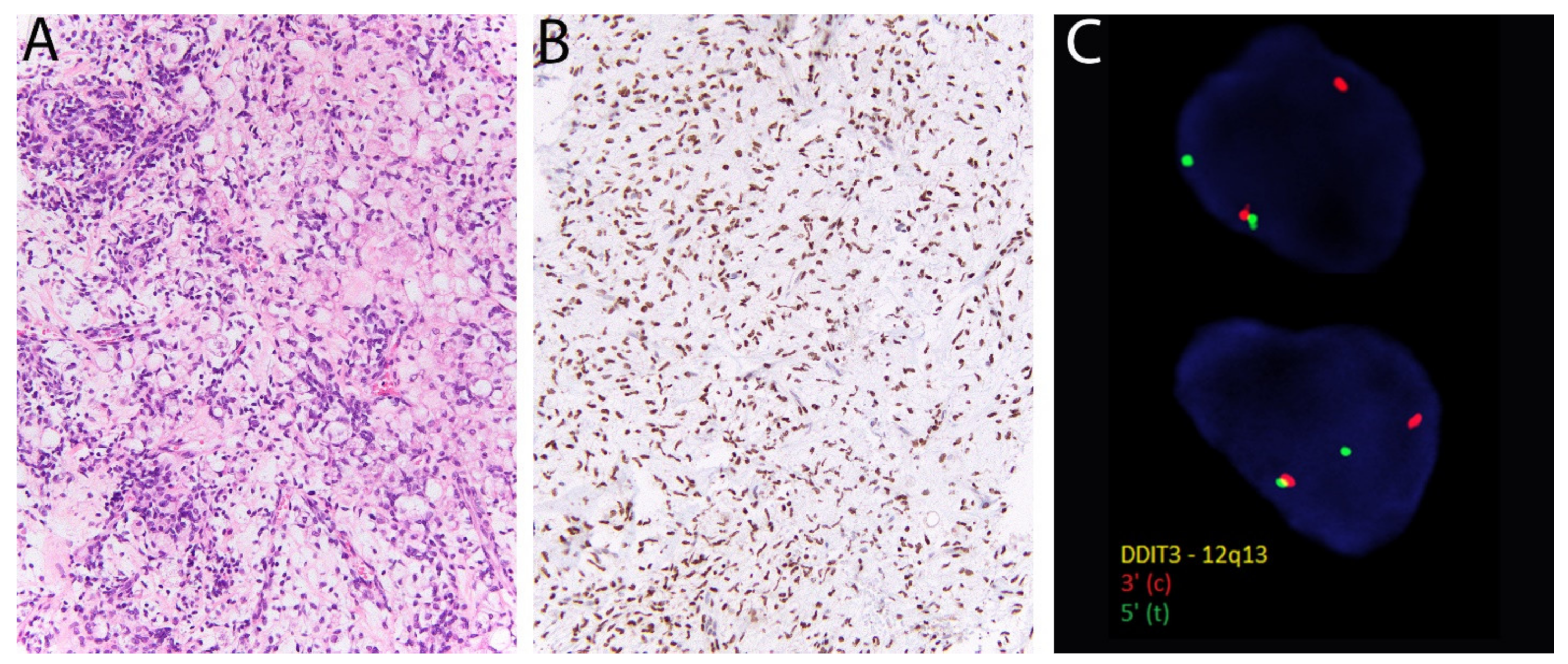
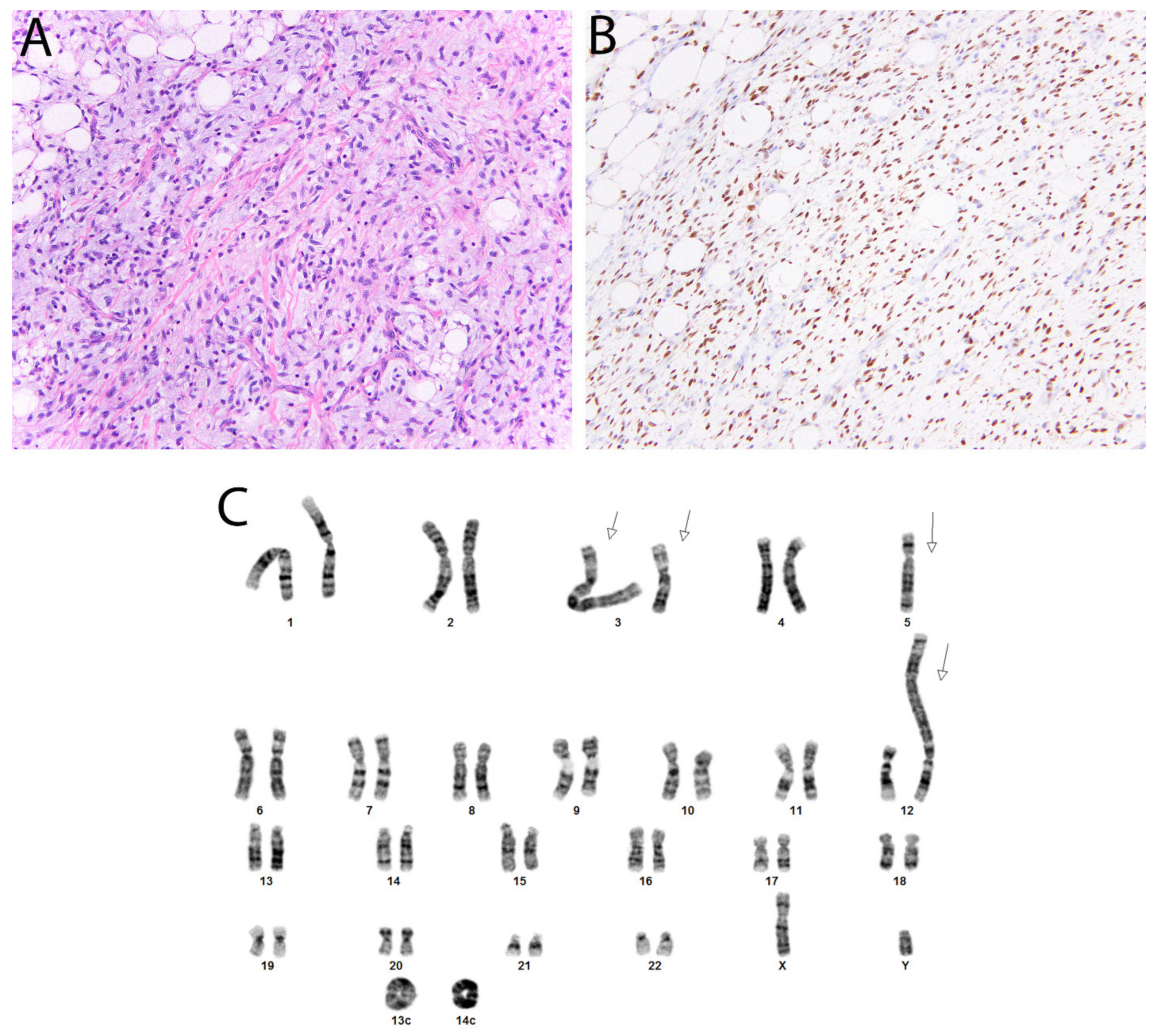
2.13. DDIT3 in Myxoid Liposarcoma
3. Immunohistochemical Markers of Gene Amplification or Inactivation
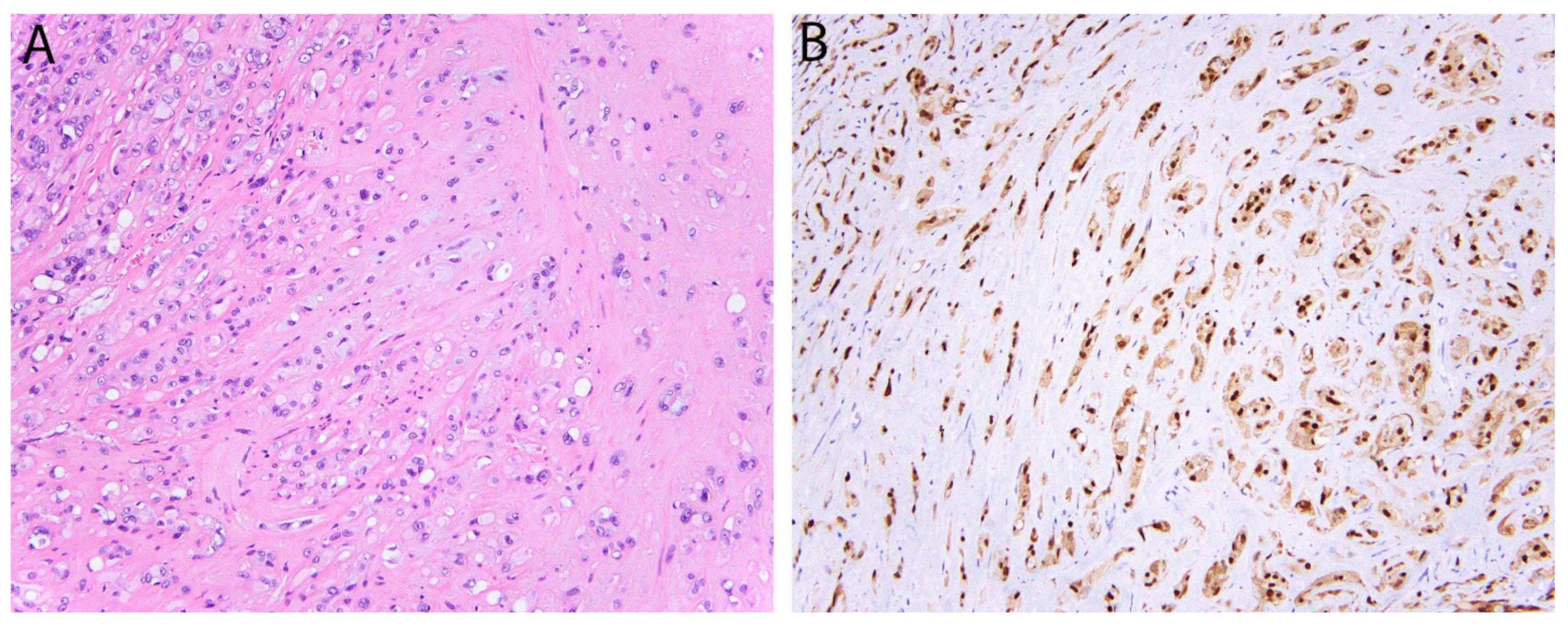
3.1. MDM2 and CDK4 in Atypical Lipomatous Tumor/Well Differentiated Liposarcoma and Dedifferentiated Liposarcoma
3.2. SWI/SNF: SMARCB1 (INI1) and SMARCA4 (BRG1) in Epithelioid Sarcoma and Others
3.3. SDH in SDH-Deficient Gastrointestinal Stromal Tumor
3.4. PRKAR1A in Malignant Melanotic Nerve Sheath Tumor and Others
4. Immunohistochemistry to Identify Epigenetic Alterations
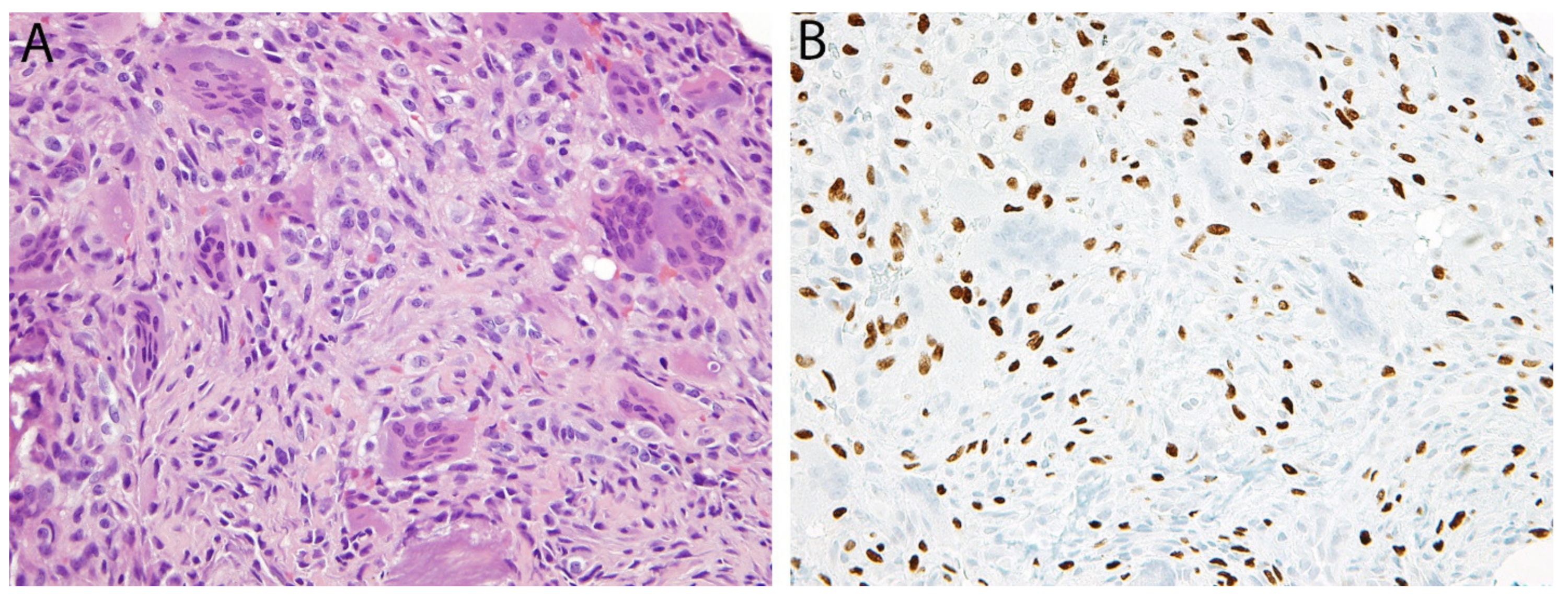
H3K27me3 in Malignant Peripheral Nerve Sheath Tumor
5. Immunohistochemical Detection of Single Nucleotide Variants
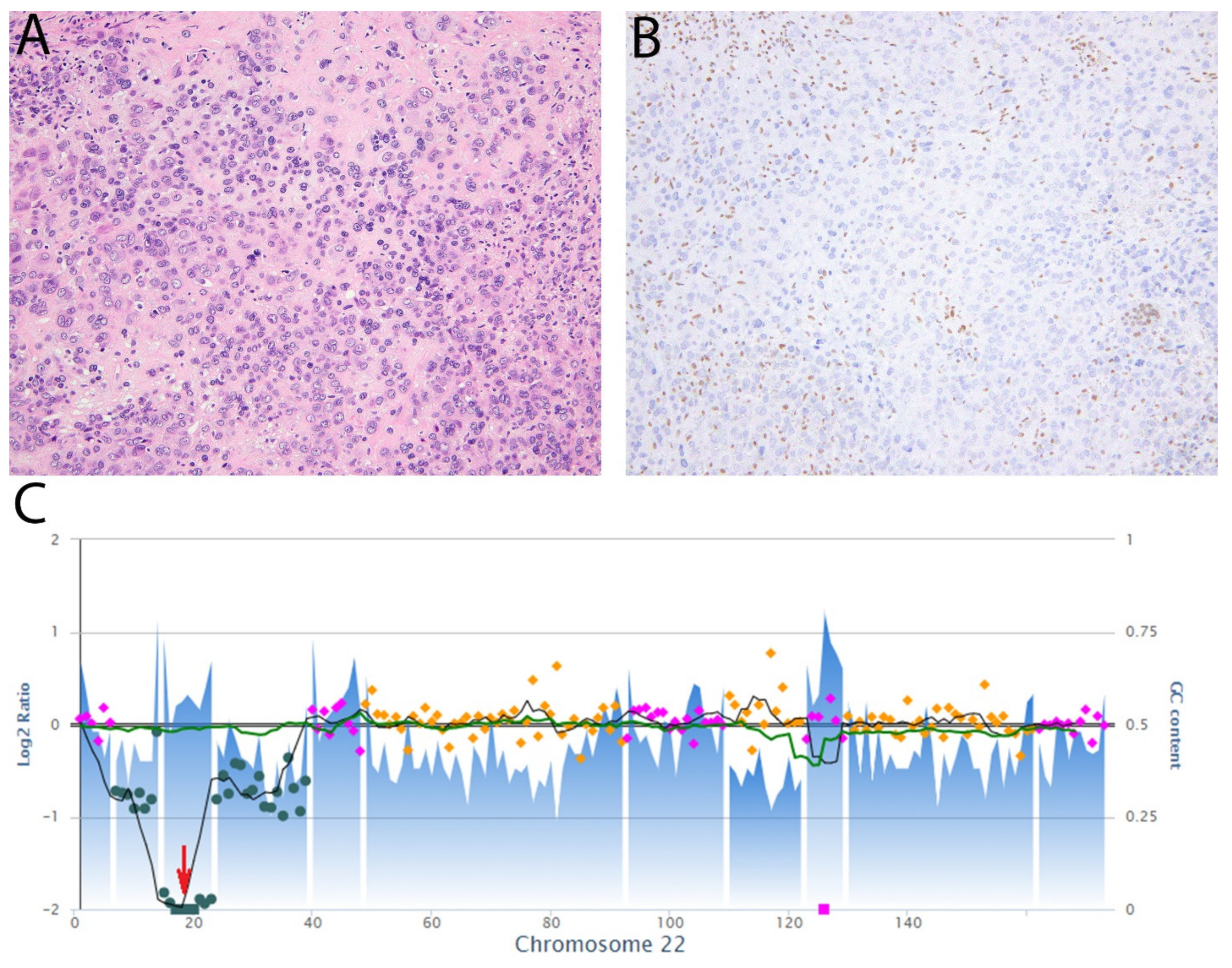
5.1. G34W in Giant Cell Tumor of Bone
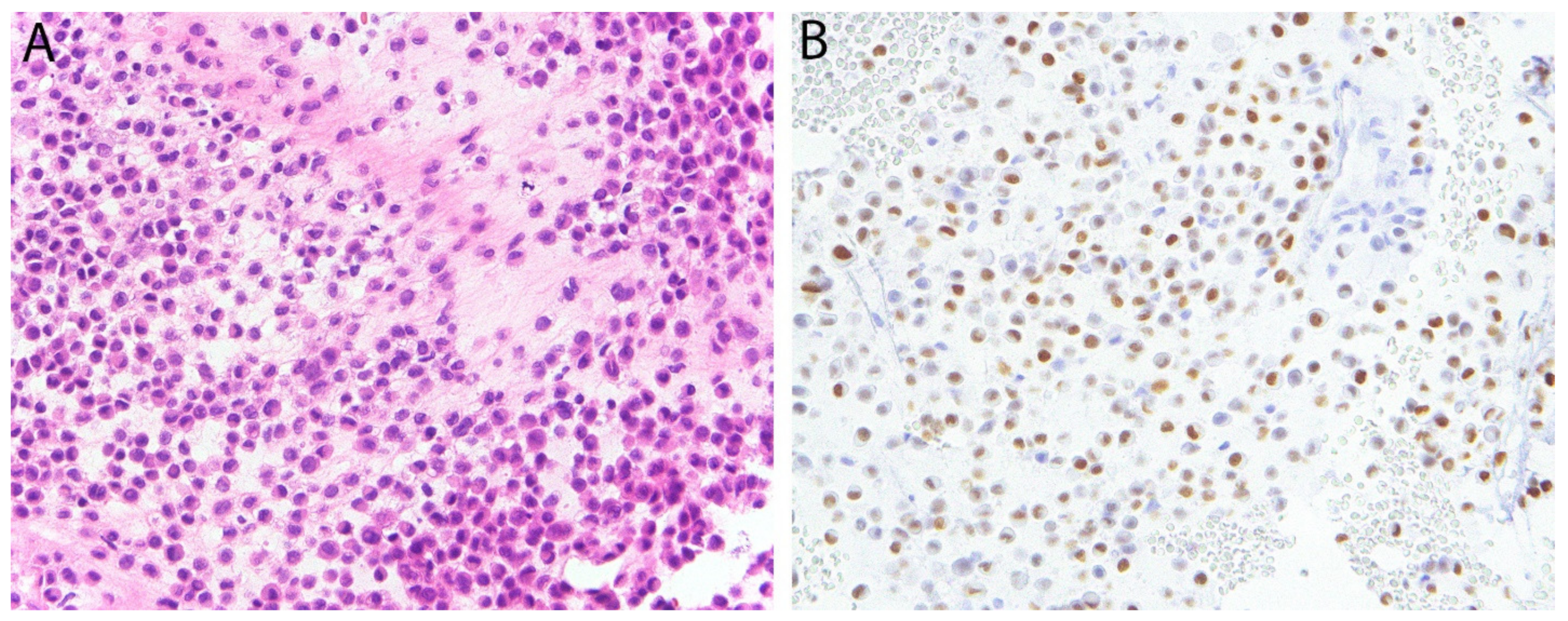
5.2. K36M in Chondroblastoma
6. Markers Discovered through Gene Expression Profiling
6.1. DOG1 in Gastrointestinal Stromal Tumor
6.2. NKX2.2 in Ewing Sarcoma and NKX3.1
6.3. ETV4 and WT1 in CIC-Rearranged Sarcoma
6.4. MUC4 in Low-Grade Fibromyxoid Sarcoma
7. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO Classification of Tumours Editorial Board. World Health Organization Classification of Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Pierron, G.; Tirode, F.; Lucchesi, C.; Reynaud, S.; Ballet, S.; Cohen-Gogo, S.; Perrin, V.; Coindre, J.M.; Delattre, O. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat. Genet. 2012, 44, 461–466. [Google Scholar] [CrossRef]
- Kao, Y.C.; Owosho, A.A.; Sung, Y.S.; Zhang, L.; Fujisawa, Y.; Lee, J.C.; Wexler, L.; Argani, P.; Swanson, D.; Dickson, B.C.; et al. BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases with Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am. J. Surg. Pathol. 2018, 42, 604–615. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shiba, E.; Umekita, Y.; Nosaka, K.; Kamio, T.; Yanai, H.; Miyasaka, C.; Watanabe, R.; Ito, I.; Tamaki, T.; et al. Clinicopathologic Diversity of Undifferentiated Sarcoma With BCOR-CCNB3 Fusion: Analysis of 11 Cases With a Reappraisal of the Utility of Immunohistochemistry for BCOR and CCNB3. Am. J. Surg. Pathol. 2017, 41, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.C.; Sung, Y.S.; Zhang, L.; Jungbluth, A.A.; Huang, S.C.; Argani, P.; Agaram, N.P.; Zin, A.; Alaggio, R.; Antonescu, C.R. BCOR Overexpression Is a Highly Sensitive Marker in Round Cell Sarcomas with BCOR Genetic Abnormalities. Am. J. Surg. Pathol. 2016, 40, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- Gerald, W.L.; Miller, H.K.; Battifora, H.; Miettinen, M.; Silva, E.G.; Rosai, J. Intra-abdominal desmoplastic small round-cell tumor. Report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am. J. Surg. Pathol. 1991, 15, 499–513. [Google Scholar] [CrossRef] [PubMed]
- de Alava, E.; Ladanyi, M.; Rosai, J.; Gerald, W.L. Detection of chimeric transcripts in desmoplastic small round cell tumor and related developmental tumors by reverse transcriptase polymerase chain reaction. A specific diagnostic assay. Am. J. Pathol. 1995, 147, 1584–1591. [Google Scholar]
- Barnoud, R.; Sabourin, J.C.; Pasquier, D.; Ranchère, D.; Bailly, C.; Terrier-Lacombe, M.J.; Pasquier, B. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: A comparative study with other small round cell tumors. Am. J. Surg. Pathol. 2000, 24, 830–836. [Google Scholar] [CrossRef]
- Rudzinski, E.R.; Anderson, J.R.; Chi, Y.Y.; Gastier-Foster, J.M.; Astbury, C.; Barr, F.G.; Skapek, S.X.; Hawkins, D.S.; Weigel, B.J.; Pappo, A.; et al. Histology, fusion status, and outcome in metastatic rhabdomyosarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef]
- Azorsa, D.O.; Bode, P.K.; Wachtel, M.; Cheuk, A.T.C.; Meltzer, P.S.; Vokuhl, C.; Camenisch, U.; Khov, H.L.; Bode, B.; Schäfer, B.W.; et al. Immunohistochemical detection of PAX-FOXO1 fusion proteins in alveolar rhabdomyosarcoma using breakpoint specific monoclonal antibodies. Mod. Pathol. 2020. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Gibson, S.; Sebire, N.J.; Anderson, J. PAX5 expression in rhabdomyosarcoma. Am. J. Surg. Pathol. 2009, 33, 1575–1577. [Google Scholar] [CrossRef]
- Sullivan, L.M.; Atkins, K.A.; LeGallo, R.D. PAX immunoreactivity identifies alveolar rhabdomyosarcoma. Am. J. Surg. Pathol. 2009, 33, 775–780. [Google Scholar] [CrossRef]
- Lewis, J.T.; Oliveira, A.M.; Nascimento, A.G.; Schembri-Wismayer, D.; Moore, E.A.; Olsen, K.D.; Garcia, J.G.; Lonzo, M.L.; Lewis, J.E. Low-grade sinonasal sarcoma with neural and myogenic features: A clinicopathologic analysis of 28 cases. Am. J. Surg. Pathol. 2012, 36, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bledsoe, K.L.; Graham, R.P.; Asmann, Y.W.; Viswanatha, D.S.; Lewis, J.E.; Lewis, J.T.; Chou, M.M.; Yaszemski, M.J.; Jen, J.; et al. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat. Genet. 2014, 46, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Le Loarer, F.; Laffont, S.; Lesluyes, T.; Tirode, F.; Antonescu, C.; Baglin, A.C.; Delespaul, L.; Soubeyran, I.; Hostein, I.; Pérot, G.; et al. Clinicopathologic and Molecular Features of a Series of 41 Biphenotypic Sinonasal Sarcomas Expanding Their Molecular Spectrum. Am. J. Surg. Pathol. 2019, 43, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Jo, V.Y.; Marino-Enriquez, A.; Fletcher, C.D.M.; Hornick, J.L. Expression of PAX3 Distinguishes Biphenotypic Sinonasal Sarcoma From Histologic Mimics. Am. J. Surg. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Crago, A.M.; Rosenberg, M.; O’Connor, R.; Walker, S.R.; Ambrogio, L.; Auclair, D.; McKenna, A.; Heinrich, M.C.; Frank, D.A.; et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat. Genet. 2013, 45, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Kalyana-Sundaram, S.; Cao, X.; Lonigro, R.J.; Sung, Y.S.; Chen, C.L.; Zhang, L.; Wang, R.; Su, F.; et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat. Genet. 2013, 45, 180–185. [Google Scholar] [CrossRef]
- Doyle, L.A.; Vivero, M.; Fletcher, C.D.; Mertens, F.; Hornick, J.L. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod. Pathol. 2014, 27, 390–395. [Google Scholar] [CrossRef]
- Yoshida, A.; Tsuta, K.; Ohno, M.; Yoshida, M.; Narita, Y.; Kawai, A.; Asamura, H.; Kushima, R. STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am. J. Surg. Pathol. 2014, 38, 552–559. [Google Scholar] [CrossRef]
- Schweizer, L.; Koelsche, C.; Sahm, F.; Piro, R.M.; Capper, D.; Reuss, D.E.; Pusch, S.; Habel, A.; Meyer, J.; Göck, T.; et al. Meningeal hemangiopericytoma and solitary fibrous tumors carry the NAB2-STAT6 fusion and can be diagnosed by nuclear expression of STAT6 protein. Acta Neuropathol. 2013, 125, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Tao, D.; Marino-Enriquez, A. STAT6 is amplified in a subset of dedifferentiated liposarcoma. Mod. Pathol. 2014, 27, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Hawkins, A.L.; Dvorak, C.; Henkle, C.; Ellingham, T.; Perlman, E.J. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999, 59, 2776–2780. [Google Scholar]
- Marino-Enriquez, A.; Wang, W.L.; Roy, A.; Lopez-Terrada, D.; Lazar, A.J.; Fletcher, C.D.; Coffin, C.M.; Hornick, J.L. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am. J. Surg. Pathol. 2011, 35, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Marino-Enriquez, A.; Fletcher, C.D.; Hornick, J.L. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod. Pathol. 2015, 28, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.N.; Yeh, I.; Jordan, R.C.; Wolsky, R.J.; Horvai, A.E.; McCalmont, T.H.; LeBoit, P.E. Cutaneous Non-Neural Granular Cell Tumors Harbor Recurrent ALK Gene Fusions. Am. J. Surg. Pathol. 2018, 42, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Acosta, A.M.; Demicco, E.G.; Dal Cin, P.; Hirsch, M.S.; Fletcher, C.D.M.; Jo, V.Y. Pseudosarcomatous myofibroblastic proliferations of the urinary bladder are neoplasms characterized by recurrent FN1-ALK fusions. Mod. Pathol. 2020. [Google Scholar] [CrossRef]
- Chang, K.T.E.; Tay, A.Z.E.; Kuick, C.H.; Chen, H.; Algar, E.; Taubenheim, N.; Campbell, J.; Mechinaud, F.; Campbell, M.; Super, L.; et al. ALK-positive histiocytosis: An expanded clinicopathologic spectrum and frequent presence of KIF5B-ALK fusion. Mod. Pathol. 2019, 32, 598–608. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Gorunova, L.; Lund-Iversen, M.; Lobmaier, I.; Bjerkehagen, B.; Heim, S. Recurrent fusion of the genes FN1 and ALK in gastrointestinal leiomyomas. Mod. Pathol. 2016, 29, 1415–1423. [Google Scholar] [CrossRef]
- Schöffski, P.; Sufliarsky, J.; Gelderblom, H.; Blay, J.Y.; Strauss, S.J.; Stacchiotti, S.; Rutkowski, P.; Lindner, L.H.; Leahy, M.G.; Italiano, A.; et al. Crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumours with and without anaplastic lymphoma kinase gene alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): A multicentre, single-drug, prospective, non-randomised phase 2 trial. Lancet Respir. Med. 2018, 6, 431–441. [Google Scholar] [CrossRef]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef]
- Knezevich, S.R.; McFadden, D.E.; Tao, W.; Lim, J.F.; Sorensen, P.H. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat. Genet. 1998, 18, 184–187. [Google Scholar] [CrossRef]
- Rudzinski, E.R.; Lockwood, C.M.; Stohr, B.A.; Vargas, S.O.; Sheridan, R.; Black, J.O.; Rajaram, V.; Laetsch, T.W.; Davis, J.L. Pan-Trk Immunohistochemistry Identifies NTRK Rearrangements in Pediatric Mesenchymal Tumors. Am. J. Surg. Pathol. 2018, 42, 927–935. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fletcher, C.D.M.; Hornick, J.L. Evaluation of pan-TRK immunohistochemistry in infantile fibrosarcoma, lipofibromatosis-like neural tumour and histological mimics. Histopathology 2018. [Google Scholar] [CrossRef]
- Bielack, S.S.; Cox, M.C.; Nathrath, M.; Apel, K.; Blattmann, C.; Holl, T.; Jenewein, R.; Klenk, U.; Klothaki, P.; Müller-Abt, P.; et al. Rapid, complete and sustained tumour response to the TRK inhibitor larotrectinib in an infant with recurrent, chemotherapy-refractory infantile fibrosarcoma carrying the characteristic ETV6-NTRK3 gene fusion. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2019, 30, viii31–viii35. [Google Scholar] [CrossRef]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Perret, R.; Velasco, V.; Le Guellec, S.; Coindre, J.M.; Le Loarer, F. The SS18-SSX Antibody Has Perfect Specificity for the SS18-SSX Fusion Protein: A Validation Study of 609 Neoplasms Including 2 Unclassified Tumors With SS18-Non-SSX Fusions. Am. J. Surg. Pathol. 2020. [Google Scholar] [CrossRef]
- Zaborowski, M.; Vargas, A.C.; Pulvers, J.; Clarkson, A.; de Guzman, D.; Sioson, L.; Maclean, F.; Chou, A.; Gill, A.J. When used together SS18-SSX fusion-specific and SSX C-terminus immunohistochemistry are highly specific and sensitive for the diagnosis of synovial sarcoma and can replace FISH or molecular testing in most cases. Histopathology 2020, 77, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.W.; Enzinger, F.M. Epithelioid hemangioendothelioma: A vascular tumor often mistaken for a carcinoma. Cancer 1982, 50, 970–981. [Google Scholar] [CrossRef]
- Errani, C.; Zhang, L.; Sung, Y.S.; Hajdu, M.; Singer, S.; Maki, R.G.; Healey, J.H.; Antonescu, C.R. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromos. Cancer 2011, 50, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Fletcher, C.D.; Hornick, J.L. Nuclear Expression of CAMTA1 Distinguishes Epithelioid Hemangioendothelioma From Histologic Mimics. Am. J. Surg. Pathol. 2016, 40, 94–102. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Le Loarer, F.; Mosquera, J.M.; Sboner, A.; Zhang, L.; Chen, C.L.; Chen, H.W.; Pathan, N.; Krausz, T.; Dickson, B.C.; et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromos. Cancer 2013, 52, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M.; Lui, M.Y.; Antonescu, C.R.; Krause-Boehm, A.; Meindl, A.; Argani, P.; Healey, J.H.; Ueda, T.; Yoshikawa, H.; Meloni-Ehrig, A.; et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 2001, 20, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Aulmann, S.; Illei, P.B.; Netto, G.J.; Ro, J.; Cho, H.Y.; Dogan, S.; Ladanyi, M.; Martignoni, G.; Goldblum, J.R.; et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am. J. Surg. Pathol. 2010, 34, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Mirra, J.M.; Kessler, S.; Bhuta, S.; Eckardt, J. The fibroma-like variant of epithelioid sarcoma. A fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer 1992, 69, 1382–1395. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Pseudomyogenic hemangioendothelioma: A distinctive, often multicentric tumor with indolent behavior. Am. J. Surg. Pathol. 2011, 35, 190–201. [Google Scholar] [CrossRef]
- Walther, C.; Tayebwa, J.; Lilljebjorn, H.; Magnusson, L.; Nilsson, J.; von Steyern, F.V.; Ora, I.; Domanski, H.A.; Fioretos, T.; Nord, K.H.; et al. A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J. Pathol. 2014, 232, 534–540. [Google Scholar] [CrossRef]
- Agaram, N.P.; Zhang, L.; Cotzia, P.; Antonescu, C.R. Expanding the Spectrum of Genetic Alterations in Pseudomyogenic Hemangioendothelioma With Recurrent Novel ACTB-FOSB Gene Fusions. Am. J. Surg. Pathol. 2018, 42, 1653–1661. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Lobmaier, I.; Gorunova, L.; Heim, S. Fusion of the Genes WWTR1 and FOSB in Pseudomyogenic Hemangioendothelioma. Cancer Genom. Proteom. 2019, 16, 293–298. [Google Scholar] [CrossRef]
- Bridge, J.A.; Sumegi, J.; Royce, T.; Baker, M.; Linos, K. A novel CLTC-FOSB gene fusion in pseudomyogenic hemangioendothelioma of bone. Genes Chromos. Cancer 2021, 60, 38–42. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. FOSB is a Useful Diagnostic Marker for Pseudomyogenic Hemangioendothelioma. Am. J. Surg. Pathol. 2017, 41, 596–606. [Google Scholar] [CrossRef]
- Huang, S.C.; Zhang, L.; Sung, Y.S.; Chen, C.L.; Krausz, T.; Dickson, B.C.; Kao, Y.C.; Agaram, N.P.; Fletcher, C.D.; Antonescu, C.R. Frequent FOS Gene Rearrangements in Epithelioid Hemangioma: A Molecular Study of 58 Cases With Morphologic Reappraisal. Am. J. Surg. Pathol. 2015, 39, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Fittall, M.W.; Mifsud, W.; Pillay, N.; Ye, H.; Strobl, A.C.; Verfaillie, A.; Demeulemeester, J.; Zhang, L.; Berisha, F.; Tarabichi, M.; et al. Recurrent rearrangements of FOS and FOSB define osteoblastoma. Nat. Commun. 2018, 9, 2150. [Google Scholar] [CrossRef] [PubMed]
- Amary, F.; Markert, E.; Berisha, F.; Ye, H.; Gerrand, C.; Cool, P.; Tirabosco, R.; Lindsay, D.; Pillay, N.; O’Donnell, P.; et al. FOS Expression in Osteoid Osteoma and Osteoblastoma: A Valuable Ancillary Diagnostic Tool. Am. J. Surg. Pathol. 2019, 43, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Scapa, J.V.; Cloutier, J.M.; Raghavan, S.S.; Peters-Schulze, G.; Varma, S.; Charville, G.W. DDIT3 Immunohistochemistry Is a Useful Tool for the Diagnosis of Myxoid Liposarcoma. Am. J. Surg. Pathol. 2020. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef]
- Hornick, J.L.; Dal Cin, P.; Fletcher, C.D. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am. J. Surg. Pathol. 2009, 33, 542–550. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Thomas, C.; Hovestadt, V.; Schrimpf, D.; Johann, P.; Bens, S.; Oyen, F.; Peetz-Dienhart, S.; Crede, Y.; Wefers, A.; et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: A distinct molecular entity with dismal prognosis. Acta Neuropathol. 2016, 132, 149–151. [Google Scholar] [CrossRef]
- Kohashi, K.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Oshiro, Y.; Izumi, T.; Taguchi, T.; Tsuneyoshi, M. SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: A special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am. J. Surg. Pathol. 2008, 32, 1168–1174. [Google Scholar] [CrossRef]
- Gleason, B.C.; Fletcher, C.D. Myoepithelial carcinoma of soft tissue in children: An aggressive neoplasm analyzed in a series of 29 cases. Am. J. Surg. Pathol. 2007, 31, 1813–1824. [Google Scholar] [CrossRef]
- Kolin, D.L.; Dong, F.; Baltay, M.; Lindeman, N.; MacConaill, L.; Nucci, M.R.; Crum, C.P.; Howitt, B.E. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): A clinicopathologic entity distinct from undifferentiated carcinoma. Mod. Pathol. 2018, 31, 1442–1456. [Google Scholar] [CrossRef] [PubMed]
- Rooper, L.M.; Uddin, N.; Gagan, J.; Brosens, L.A.A.; Magliocca, K.R.; Edgar, M.A.; Thompson, L.D.R.; Agaimy, A.; Bishop, J.A. Recurrent Loss of SMARCA4 in Sinonasal Teratocarcinosarcoma. Am. J. Surg. Pathol. 2020, 44, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Kohashi, K.; Yamamoto, H.; Yamada, Y.; Kinoshita, I.; Taguchi, T.; Iwamoto, Y.; Oda, Y. SWI/SNF Chromatin-remodeling Complex Status in SMARCB1/INI1-preserved Epithelioid Sarcoma. Am. J. Surg. Pathol. 2018, 42, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Rekhtman, N.; Montecalvo, J.; Chang, J.C.; Alex, D.; Ptashkin, R.N.; Ai, N.; Sauter, J.L.; Kezlarian, B.; Jungbluth, A.; Desmeules, P.; et al. SMARCA4-Deficient Thoracic Sarcomatoid Tumors Represent Primarily Smoking-Related Undifferentiated Carcinomas Rather Than Primary Thoracic Sarcomas. J. Thorac. Oncol. 2020, 15, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Remillard, S.P.; Zhang, Y.X.; Doyle, L.A.; George, S.; Hornick, J.L. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod. Pathol. 2013, 26, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Torres-Mora, J.; Dry, S.; Li, X.; Binder, S.; Amin, M.; Folpe, A.L. Malignant melanotic schwannian tumor: A clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am. J. Surg. Pathol. 2014, 38, 94–105. [Google Scholar] [CrossRef]
- Wang, L.; Zehir, A.; Sadowska, J.; Zhou, N.; Rosenblum, M.; Busam, K.; Agaram, N.; Travis, W.; Arcila, M.; Dogan, S.; et al. Consistent copy number changes and recurrent PRKAR1A mutations distinguish Melanotic Schwannomas from Melanomas: SNP-array and next generation sequencing analysis. Genes Chromos. Cancer 2015, 54, 463–471. [Google Scholar] [CrossRef]
- Maleszewski, J.J.; Larsen, B.T.; Kip, N.S.; Castonguay, M.C.; Edwards, W.D.; Carney, J.A.; Kipp, B.R. PRKAR1A in the development of cardiac myxoma: A study of 110 cases including isolated and syndromic tumors. Am. J. Surg. Pathol. 2014, 38, 1079–1087. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Fletcher, C.D.; Hornick, J.L. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod. Pathol. 2016, 29, 4–13. [Google Scholar] [CrossRef]
- Cleven, A.H.; Sannaa, G.A.; Briaire-de Bruijn, I.; Ingram, D.R.; van de Rijn, M.; Rubin, B.P.; de Vries, M.W.; Watson, K.L.; Torres, K.E.; Wang, W.L.; et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016, 29, 582–590. [Google Scholar] [CrossRef]
- Prieto-Granada, C.N.; Wiesner, T.; Messina, J.L.; Jungbluth, A.A.; Chi, P.; Antonescu, C.R. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am. J. Surg. Pathol. 2016, 40, 479–489. [Google Scholar] [CrossRef]
- Lyskjaer, I.; Lindsay, D.; Tirabosco, R.; Steele, C.D.; Lombard, P.; Strobl, A.C.; Rocha, A.M.; Davies, C.; Ye, H.; Bekers, E.; et al. H3K27me3 expression and methylation status in histological variants of malignant peripheral nerve sheath tumours. J. Pathol. 2020, 252, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Panse, G.; Mito, J.K.; Ingram, D.R.; Wani, K.; Khan, S.; Lazar, A.J.; Doyle, L.A.; Wang, W.L. Radiation-associated sarcomas other than malignant peripheral nerve sheath tumours demonstrate loss of histone H3K27 trimethylation(†). Histopathology 2020. [Google Scholar] [CrossRef]
- Le Guellec, S.; Macagno, N.; Velasco, V.; Lamant, L.; Lae, M.; Filleron, T.; Malissen, N.; Cassagnau, E.; Terrier, P.; Chevreau, C.; et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: A study of 387 cases including mimicking lesions. Mod. Pathol. 2017, 30, 1677–1687. [Google Scholar] [CrossRef]
- Marchione, D.M.; Lisby, A.; Viaene, A.N.; Santi, M.; Nasrallah, M.; Wang, L.P.; Williams, E.A.; Larque, A.B.; Chebib, I.; Garcia, B.A.; et al. Histone H3K27 dimethyl loss is highly specific for malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss. Mod. Pathol. 2019, 32, 1434–1446. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Presneau, N.; Baumhoer, D.; Behjati, S.; Pillay, N.; Tarpey, P.; Campbell, P.J.; Jundt, G.; Hamoudi, R.; Wedge, D.C.; Loo, P.V.; et al. Diagnostic value of H3F3A mutations in giant cell tumour of bone compared to osteoclast-rich mimics. J. Pathol. Clin. Res. 2015, 1, 113–123. [Google Scholar] [CrossRef]
- Amary, F.; Berisha, F.; Ye, H.; Gupta, M.; Gutteridge, A.; Baumhoer, D.; Gibbons, R.; Tirabosco, R.; O’Donnell, P.; Flanagan, A.M. H3F3A (Histone 3.3) G34W Immunohistochemistry: A Reliable Marker Defining Benign and Malignant Giant Cell Tumor of Bone. Am. J. Surg. Pathol. 2017, 41, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Berisha, F.; Mozela, R.; Gibbons, R.; Guttridge, A.; O’Donnell, P.; Baumhoer, D.; Tirabosco, R.; Flanagan, A.M. The H3F3 K36M mutant antibody is a sensitive and specific marker for the diagnosis of chondroblastoma. Histopathology 2016, 69, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.H.; Srivastava, A.; Kim, Y.E.; Park, H.J.; Park, C.K.; Sohn, T.S.; Kim, S.; Kang, D.Y.; Kim, K.M. DOG1 and PKC-θ are useful in the diagnosis of KIT-negative gastrointestinal stromal tumors. Mod. Pathol. 2011, 24, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; West, R.B.; Linn, S.C.; Alter, O.; Knowling, M.A.; O’Connell, J.X.; Zhu, S.; Fero, M.; Sherlock, G.; Pollack, J.R.; et al. Molecular characterisation of soft tissue tumours: A gene expression study. Lancet 2002, 359, 1301–1307. [Google Scholar] [CrossRef]
- West, R.B.; Corless, C.L.; Chen, X.; Rubin, B.P.; Subramanian, S.; Montgomery, K.; Zhu, S.; Ball, C.A.; Nielsen, T.O.; Patel, R.; et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am. J. Pathol. 2004, 165, 107–113. [Google Scholar] [CrossRef]
- Miettinen, M.; Wang, Z.F.; Lasota, J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: A study of 1840 cases. Am. J. Surg. Pathol. 2009, 33, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, F.; Corless, C.L.; Duensing, A.; Hornick, J.L.; Oliveira, A.M.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D. KIT-negative gastrointestinal stromal tumors: Proof of concept and therapeutic implications. Am. J. Surg. Pathol. 2004, 28, 889–894. [Google Scholar] [CrossRef]
- Agaimy, A.; Otto, C.; Braun, A.; Geddert, H.; Schaefer, I.M.; Haller, F. Value of epithelioid morphology and PDGFRA immunostaining pattern for prediction of PDGFRA mutated genotype in gastrointestinal stromal tumors (GISTs). Int. J. Clin. Exp. Pathol. 2013, 6, 1839–1846. [Google Scholar] [PubMed]
- DeMatteo, R.P.; Ballman, K.V.; Antonescu, C.R.; Corless, C.; Kolesnikova, V.; von Mehren, M.; McCarter, M.D.; Norton, J.; Maki, R.G.; Pisters, P.W.; et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor: ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann. Surg. 2013, 258, 422–429. [Google Scholar] [CrossRef]
- Dematteo, R.P.; Ballman, K.V.; Antonescu, C.R.; Maki, R.G.; Pisters, P.W.; Demetri, G.D.; Blackstein, M.E.; Blanke, C.D.; von Mehren, M.; Brennan, M.F.; et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 1097–1104. [Google Scholar] [CrossRef]
- Briscoe, J.; Sussel, L.; Serup, P.; Hartigan-O’Connor, D.; Jessell, T.M.; Rubenstein, J.L.; Ericson, J. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature 1999, 398, 622–627. [Google Scholar] [CrossRef]
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006, 9, 405–416. [Google Scholar] [CrossRef]
- Yoshida, A.; Sekine, S.; Tsuta, K.; Fukayama, M.; Furuta, K.; Tsuda, H. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am. J. Surg. Pathol. 2012, 36, 993–999. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: Imperfect specificity for Ewing sarcoma. Mod. Pathol. 2016, 29, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.I.; Machado, I.; Motoi, T.; Parafioriti, A.; Lacambra, M.; Ichikawa, H.; Kawai, A.; Antonescu, C.R.; Yoshida, A. NKX3-1 Is a Useful Immunohistochemical Marker of EWSR1-NFATC2 Sarcoma and Mesenchymal Chondrosarcoma. Am. J. Surg. Pathol. 2020, 44, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Perret, R.; Escuriol, J.; Velasco, V.; Mayeur, L.; Soubeyran, I.; Delfour, C.; Aubert, S.; Polivka, M.; Karanian, M.; Meurgey, A.; et al. NFATc2-rearranged sarcomas: Clinicopathologic, molecular, and cytogenetic study of 7 cases with evidence of AGGRECAN as a novel diagnostic marker. Mod. Pathol. 2020, 33, 1930–1944. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hornick, J.L.; Fletcher, C.D.M. NKX3.1 immunoreactivity is not identified in mesenchymal chondrosarcoma: A 25-case cohort study. Histopathology 2021, 78, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Hashimoto, T.; Ryo, E.; Yoshida, K.I.; Motoi, T.; Yatabe, Y.; Mori, T. Confirmation of NKX3-1 Expression in EWSR1-NFATC2 Sarcoma and Mesenchymal Chondrosarcoma Using Monoclonal Antibody Immunohistochemistry, RT-PCR, and RNA In Situ Hybridization. Am. J. Surg. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Owosho, A.A.; Zhang, L.; Chen, S.; Deniz, K.; Huryn, J.M.; Kao, Y.C.; Huang, S.C.; Singer, S.; Tap, W.; et al. Sarcomas With CIC-rearrangements Are a Distinct Pathologic Entity With Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am. J. Surg. Pathol. 2017, 41, 941–949. [Google Scholar] [CrossRef]
- Italiano, A.; Sung, Y.S.; Zhang, L.; Singer, S.; Maki, R.G.; Coindre, J.M.; Antonescu, C.R. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromos. Cancer 2012, 51, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of ETV4 and WT1 expression in CIC-rearranged sarcomas and histologic mimics. Mod. Pathol. 2016, 29, 1324–1334. [Google Scholar] [CrossRef]
- Siegele, B.; Roberts, J.; Black, J.O.; Rudzinski, E.; Vargas, S.O.; Galambos, C. DUX4 Immunohistochemistry Is a Highly Sensitive and Specific Marker for CIC-DUX4 Fusion-positive Round Cell Tumor. Am. J. Surg. Pathol. 2017, 41, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Möller, E.; Hornick, J.L.; Magnusson, L.; Veerla, S.; Domanski, H.A.; Mertens, F. FUS-CREB3L2/L1-positive sarcomas show a specific gene expression profile with upregulation of CD24 and FOXL1. Clin. Cancer Res. 2011, 17, 2646–2656. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L. Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having a deceptively benign appearance. Am. J. Clin. Pathol. 1987, 88, 615–619. [Google Scholar] [CrossRef]
- Doyle, L.A.; Wang, W.L.; Dal Cin, P.; Lopez-Terrada, D.; Mertens, F.; Lazar, A.J.; Fletcher, C.D.; Hornick, J.L. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: Association with FUS gene rearrangement. Am. J. Surg. Pathol. 2012, 36, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Möller, E.; Dal Cin, P.; Fletcher, C.D.; Mertens, F.; Hornick, J.L. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am. J. Surg. Pathol. 2011, 35, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Forgó, E.; Hornick, J.L.; Charville, G.W. MUC4 is expressed in alveolar rhabdomyosarcoma. Histopathology 2020. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alteration | Marker | Tumor Type | Molecular Alterations |
|---|---|---|---|
| Gene fusion | CCNB3 | BCOR-CCNB3 sarcoma | BCOR-CCNB3 fusion |
| BCOR | BCOR-CCNB3 sarcoma | BCOR-CCNB3 fusion | |
| Primitive myxoid mesenchymal tumor of infancy | BCOR internal tandem duplications | ||
| Clear cell sarcoma of kidney | BCOR internal tandem duplications | ||
| WT1 C-terminus | Desmoplastic small round cell tumor | EWSR1-WT1 fusion | |
| PAX3/7-FOXO1 | Alveolar rhabdomyosarcoma | PAX3-FOXO1 fusion (~80%) PAX7-FOXO1 fusion (~20%) | |
| PAX3 | Biphenotypic sinonasal sarcoma | PAX3 fusions | |
| STAT6 | Solitary fibrous tumor | NAB2-STAT6 fusion | |
| ALK | Inflammatory myofibroblastic tumor | ALK fusions (~60%) | |
| ROS1 | Inflammatory myofibroblastic tumor | ROS1 fusions (<10%) | |
| Pan-Trk | Infantile fibrosarcoma | ETV6-NTRK3 fusion | |
| NTRK-rearranged spindle cell neoplasms | NTRK1/2/3 fusions | ||
| SS18-SSX | Synovial sarcoma | SS18-SSX1/2/4 fusion | |
| CAMTA1 | Epithelioid hemangioendothelioma | WWTR1-CAMTA1 fusion (>90%) | |
| TFE3 | Alveolar soft part sarcoma | ASPSCR1-TFE3 fusion | |
| Epithelioid hemangioendothelioma | YAP1-TFE3 fusion (<10%) | ||
| PEComa | TFE3 fusions, TSC1/TSC2 inactivation | ||
| FOSB | Pseudomyogenic hemangioendothelioma | FOSB fusions | |
| Epithelioid hemangioma | FOSB/FOS fusions (~50%) | ||
| FOS | Osteoblastoma and osteoid osteoma | FOS rearrangements (~90%) | |
| DDIT3 | Myxoid liposarcoma | FUS-DDIT3 fusion | |
| Amplification | MDM2/CDK4 | Well-differentiated liposarcoma /Atypical lipomatous tumor | 12q13-15 (MDM2/CDK4) amplification |
| Intimal sarcoma | 12q13-15 (MDM2/CDK4) amplification | ||
| Low-grade central osteosarcoma and parosteal osteosarcoma | 12q13-15 (MDM2/CDK4) amplification | ||
| Inactivation | SMARCB1 | Epithelioid sarcoma | SMARCB1 inactivation |
| Malignant rhabdoid tumor | SMARCB1 inactivation | ||
| Epithelioid malignant peripheral nerve sheath tumor | SMARCB1 inactivation | ||
| Poorly differentiated chordoma | SMARCB1 inactivation | ||
| SMARCA4 | Thoracic SMARCA4-deficient undifferentiated tumor | SMARCA4 inactivation | |
| SMARCA4-deficient uterine sarcoma | SMARCA4 inactivation | ||
| SDHB | Gastrointestinal stromal tumor | SDH inactivation | |
| PRKAR1A | Malignant melanotic nerve sheath tumor | PRKAR1A inactivation | |
| Epigenetic | H3K27me3 | Malignant peripheral nerve sheath tumor | NF1 inactivation, SUZ12 or EED inactivation |
| SNV | G34W | Giant cell tumor of bone | H3-3A (H3F3A) mutations: p.Gly34Trp (G34W) ~90% |
| K36M | Chondroblastoma | H3-3B (H3F3B) mutations: p.Lys36Met (K36M) ~95% | |
| Overexpression | NKX2.2 | Ewing sarcoma | EWSR1-FLI1 (85%) EWSR1-ERG (10%) EWSR1-ETS gene FUS-ETS gene |
| NKX3.1 | Mesenchymal chondrosarcoma | HEY1-NCOA2 fusion (~100%) | |
| EWSR1-NFATC2 and FUS-NFATC2 sarcomas | EWSR1-NFATC2 and FUS-NFATC2 fusions | ||
| WT1 and ETV4 | CIC-rearranged sarcomas | CIC-DUX4 (95%) CIC-NUTM1, CIC-FOXO4, CIC-LEUTX | |
| MUC4 | Low grade fibromyxoid sarcoma sclerosing epithelioid fibrosarcoma | FUS-CREB3L2 fusion FUS-CREB3L1 fusion | |
| DOG1 | Gastrointestinal stromal tumor | KIT, PDGFRA, SDH, BRAF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, W.J.; Jo, V.Y. Diagnostic Immunohistochemistry of Soft Tissue and Bone Tumors: An Update on Biomarkers That Correlate with Molecular Alterations. Diagnostics 2021, 11, 690. https://doi.org/10.3390/diagnostics11040690
Anderson WJ, Jo VY. Diagnostic Immunohistochemistry of Soft Tissue and Bone Tumors: An Update on Biomarkers That Correlate with Molecular Alterations. Diagnostics. 2021; 11(4):690. https://doi.org/10.3390/diagnostics11040690
Chicago/Turabian StyleAnderson, William J., and Vickie Y. Jo. 2021. "Diagnostic Immunohistochemistry of Soft Tissue and Bone Tumors: An Update on Biomarkers That Correlate with Molecular Alterations" Diagnostics 11, no. 4: 690. https://doi.org/10.3390/diagnostics11040690
APA StyleAnderson, W. J., & Jo, V. Y. (2021). Diagnostic Immunohistochemistry of Soft Tissue and Bone Tumors: An Update on Biomarkers That Correlate with Molecular Alterations. Diagnostics, 11(4), 690. https://doi.org/10.3390/diagnostics11040690