
Prenatal Exome Sequencing: Background, Current Practice and Future Perspectives—A Systematic Review

 ,
,
Abstract
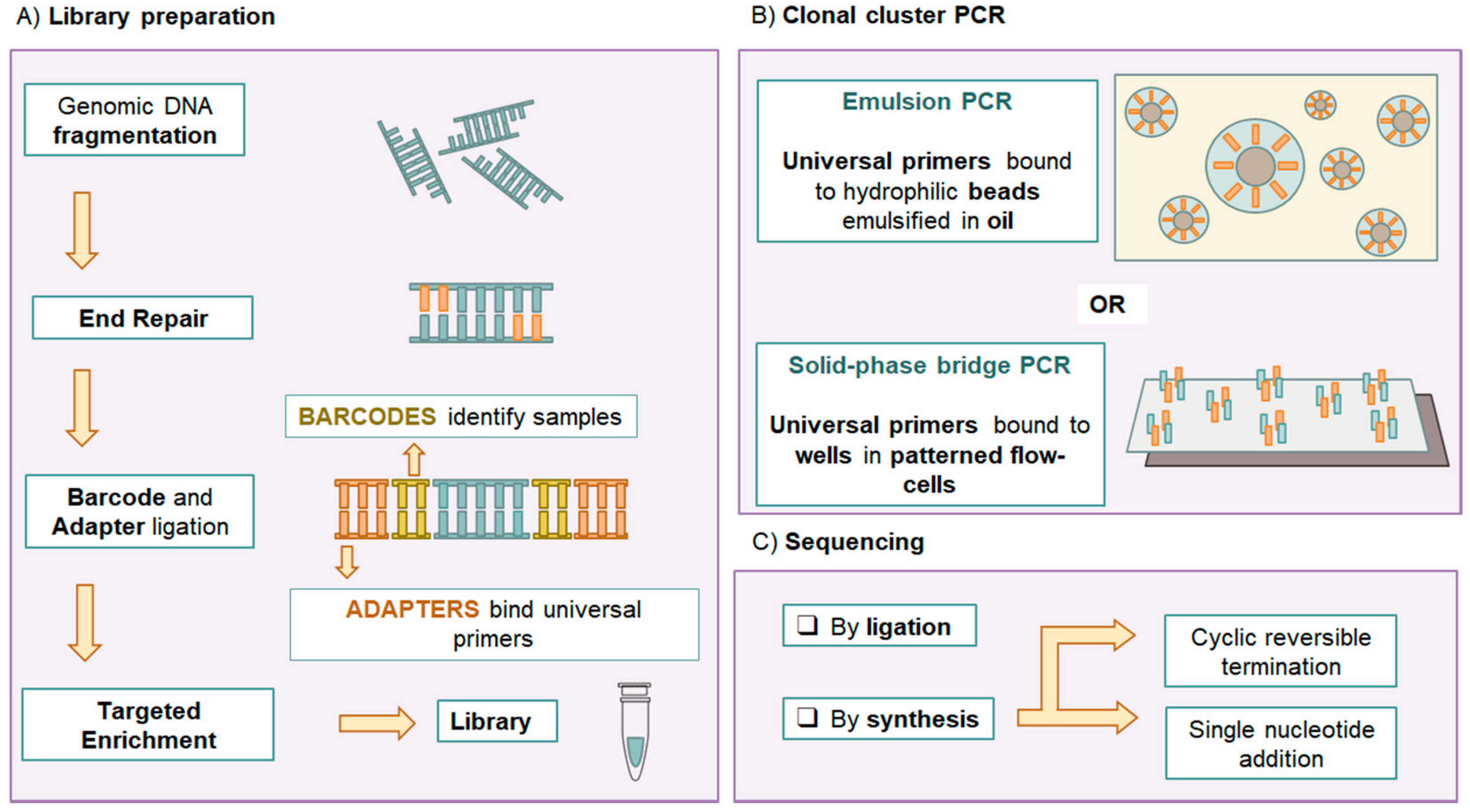
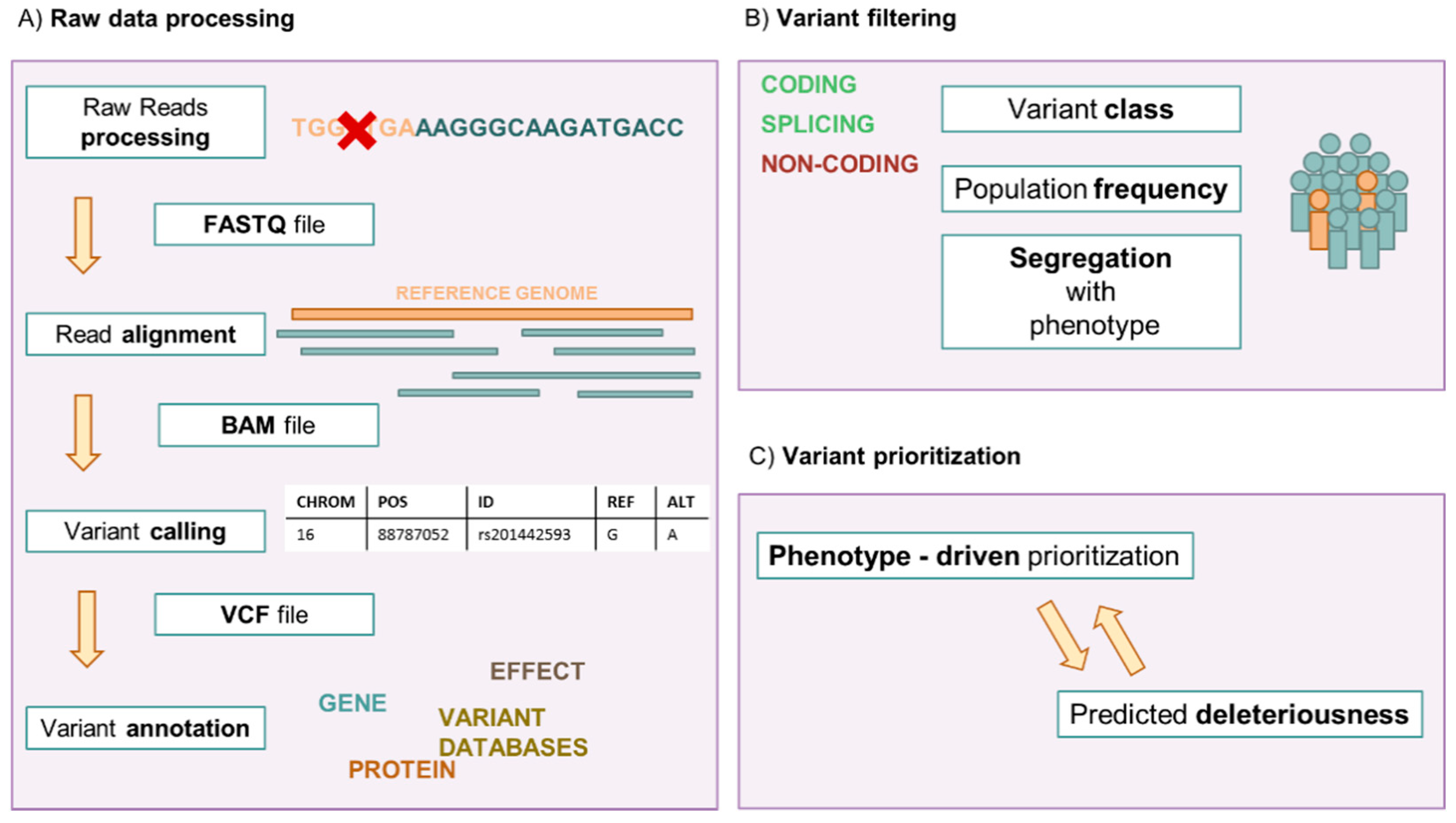
1. Introduction
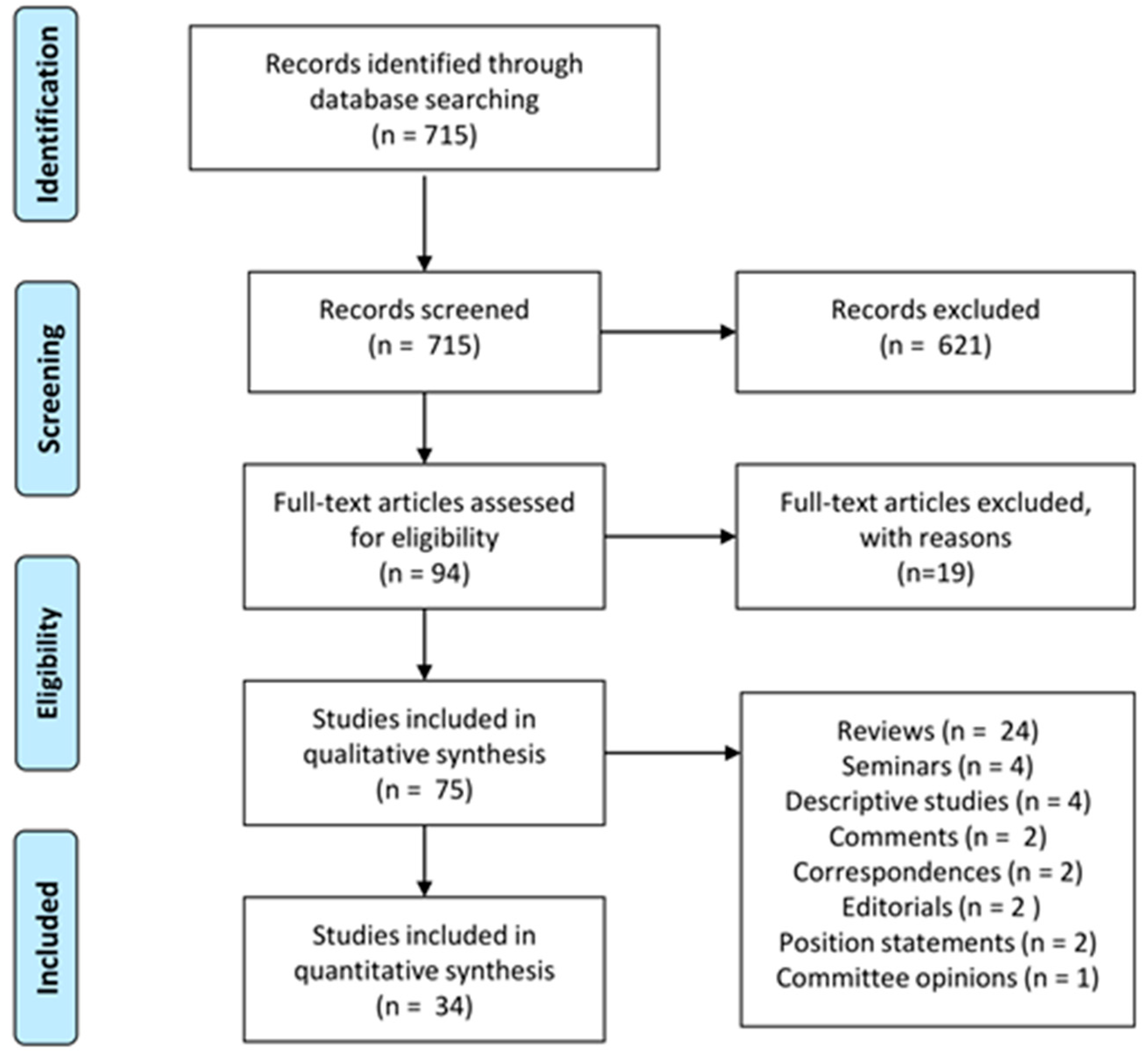
2. Materials and Methods
3. Results
3.1. pES in Fetuses Selected for US Anomalies (Regardless of the Affected Organ)
3.2. pES in Fetuses Selected for Specific Class Anomalies
3.3. Other Studies
- 14-case study due to lack of inclusion criteria [62].
- 7-case study due to lack of clear eligibility criteria [63].
- 44-case study due to the higher a-priori risk for consanguinity and recurrence [64].
- 80-families study, in which they tested parents for recessive disorders [65].
- 73-samples study, because they did not present sufficient data to be compared to the other papers [66].
- 20-case study because it combined prenatal and postnatal phenotyping to interpret WES variants [67].
- 45-case study, Jewish descent, excluded due to the different inclusion criteria and the ethnicity at high risk for recessive disorders [68].
- 19-case study, for inhomogeneity in inclusion criteria and chromosomal anomalies/CNV assessment [69].
- 6-case study for inhomogeneity in inclusion criteria and chromosomal anomalies/CNV assessment [70].
- 102-case study, because 15 fetuses were elected for multiple anomalies highly suggestive of a genetic disorder, while further enrollment was extended to each pregnancy with fetal anomaly [71].
- 183-case study because it was designed to identify novel genes causing CAKUT [72].
- 30-case study, as the same cases were also included in a subsequent study [73].
- 56-case study, because pES was performed after a negative gene panel [74].
- 9-case study due to the unsystematic pES accession [75].
- 68-case study because they proposed a diagnostic algorithm for the Bardet-Biedl syndrome diagnosis, without presenting cases [76].
- 16-case study because they performed panel genes [77].
- 6-case study because it investigated a very specific phenotype after negative panel [78].
- 708-case study due to the postnatal diagnosis [79].
4. Discussion
4.1. pES Cohorts and Series Analysis
4.1.1. Exome Sequencing in Fetuses Enrolled by US Anomalies (Regardless of the Affected Organ)
4.1.2. Exome Sequencing in Fetuses Selected for Specific Class Anomaly
4.2. Recent Past and State of the Art
4.3. Present Challenges
4.4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Van den Veyver, I.B.; Eng, C.M. Genome-Wide Sequencing for Prenatal Detection of Fetal Single-Gene Disorders. Cold Spring Harb. Perspect Med. 2015, 5, a023077. [Google Scholar] [CrossRef]
- Wou, K.; DeBie, I.; Carroll, J.; Brock, J.A.; Douglas Wilson, R. Fetal Exome Sequencing on the Horizon. J. Obs. Gynaecol. Can. 2019, 41, 64–67. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Sun, Y.; Ruivenkamp, C.A.; Hoffer, M.J.; Vrijenhoek, T.; Kriek, M.; van Asperen, C.J.; den Dunnen, J.T.; Santen, G.W. Next-generation diagnostics: Gene panel, exome, or whole genome? Hum. Mutat. 2015, 36, 648–655. [Google Scholar] [CrossRef]
- Raffan, E.; Semple, R.K. Next generation sequencing--implications for clinical practice. Br. Med. Bull. 2011, 99, 53–71. [Google Scholar] [CrossRef]
- Pengelly, R.J.; Ward, D.; Hunt, D.; Mattocks, C.; Ennis, S. Comparison of Mendeliome exome capture kits for use in clinical diagnostics. Sci. Rep. 2020, 10, 3235. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Polev, D.E.; Glotov, A.S.; Serebryakova, E.A.; Shcherbakova, I.V.; Kiselev, A.M.; Kostareva, A.A.; Glotov, O.S.; Predeus, A.V. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep. 2020, 10, 2057. [Google Scholar] [CrossRef]
- Basran, R.K.; Marshall, C.R.; Shlien, A.; Eliou, M.; Orr, J.; Lau, L.; Stavropoulos, D.J.; Ray, P.N. MG-129 Our experience of in silico gene panel testing for clinically heterogeneous disorders using exome sequencing. J. Med. Genet. 2015, 52, A11. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Moreno-Cabrera, J.M.; del Valle, J.; Castellanos, E.; Feliubadaló, L.; Pineda, M.; Brunet, J.; Serra, E.; Capellà, G.; Lázaro, C.; Gel, B. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur. J. Hum. Genet. 2020, 28, 1645–1655. [Google Scholar] [CrossRef]
- Westerfield, L.; Darilek, S.; van den Veyver, I.B. Counseling Challenges with Variants of Uncertain Significance and Incidental Findings in Prenatal Genetic Screening and Diagnosis. J. Clin. Med. 2014, 3, 1018–1032. [Google Scholar] [CrossRef]
- Horn, R.; Parker, M. Opening Pandora’s box? Ethical issues in prenatal whole genome and exome sequencing. Prenat. Diagn. 2018, 38, 20–25. [Google Scholar] [CrossRef]
- Van den Veyver, I.B. Recent advances in prenatal genetic screening and testing. F1000Research 2016, 5, 2591. [Google Scholar] [CrossRef]
- Seo, G.H.; Kim, T.; Choi, I.H.; Park, J.Y.; Lee, J.; Kim, S.; Won, D.G.; Oh, A.; Lee, Y.; Choi, J.; et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 2020, 98, 562–570. [Google Scholar] [CrossRef]
- Mone, F.; Quinlan-Jones, E.; Kilby, M.D. Clinical utility of exome sequencing in the prenatal diagnosis of congenital anomalies: A Review. Eur. J. Obs. Gynecol. Reprod. Biol. 2018, 231, 19–24. [Google Scholar] [CrossRef]
- Krstić, N.; Običan, S.G. Current landscape of prenatal genetic screening and testing. Birth. Defects Res. 2020, 112, 321–331. [Google Scholar] [CrossRef]
- Babkina, N.; Graham, J.M., Jr. New genetic testing in prenatal diagnosis. Semin. Fetal Neonatal Med. 2014, 19, 214–219. [Google Scholar] [CrossRef]
- Hayward, J.; Chitty, L.S. Beyond screening for chromosomal abnormalities: Advances in non-invasive diagnosis of single gene disorders and fetal exome sequencing. Semin. Fetal Neonatal Med. 2018, 23, 94–101. [Google Scholar] [CrossRef]
- Hashiloni-Dolev, Y.; Nov-Klaiman, T.; Raz, A. Pandora’s pregnancy: NIPT, CMA, and genome sequencing—A new era for prenatal genetic testing. Prenat. Diagn. 2019, 39, 859–865. [Google Scholar] [CrossRef]
- Jelin, A.C.; Sagaser, K.G.; Wilkins-Haug, L. Prenatal Genetic Testing Options. Pediatr. Clin. N. Am. 2019, 66, 281–293. [Google Scholar] [CrossRef]
- Jelin, A.C.; Vora, N. Whole Exome Sequencing: Applications in Prenatal Genetics. Obs. Gynecol. Clin. N. Am. 2018, 45, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Wapner, R. Prenatal diagnosis by chromosomal microarray analysis. Fertil. Steril. 2018, 109, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Mone, F.; Quinlan-Jones, E.; Ewer, A.K.; Kilby, M.D. Exome sequencing in the assessment of congenital malformations in the fetus and neonate. Arch. Dis. Child. Fetal Neonatal Ed. 2019, 104, F452–F456. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Rehm, H.L. Introduction of genomics into prenatal diagnostics. Lancet 2019, 393, 719–721. [Google Scholar] [CrossRef]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; PRISMA-P Group. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: Elaboration and explanation. BMJ 2015, 350, g7647. [Google Scholar] [CrossRef]
- Pangalos, C.; Hagnefelt, B.; Lilakos, K.; Konialis, C. First applications of a targeted exome sequencing approach in fetuses with ultrasound abnormalities reveals an important fraction of cases with associated gene defects. PeerJ 2016, 26, e1955. [Google Scholar] [CrossRef]
- Vora, N.L.; Powell, B.; Brandt, A.; Strande, N.; Hardisty, E.; Gilmore, K.; Foreman, A.K.M.; Wilhelmsen, K.; Bizon, C.; Reilly, J.; et al. Prenatal exome sequencing in anomalous fetuses: New opportunities and challenges. Genet. Med. 2017, 19, 1207–1216. [Google Scholar] [CrossRef]
- Yates, C.L.; Monaghan, K.G.; Copenheaver, D.; Retterer, K.; Scuffins, J.; Kucera, C.R.; Friedman, B.; Richard, G.; Juusola, J. Whole-exome sequencing on deceased fetuses with ultrasound anomalies: Expanding our knowledge of genetic disease during fetal development. Genet. Med. 2017, 19, 1171–1178. [Google Scholar] [CrossRef]
- Boissel, S.; Fallet-Bianco, C.; Chitayat, D.; Kremer, V.; Nassif, C.; Rypens, F.; Delrue, M.A.; Dal Soglio, D.; Oligny, L.L.; Patey, N.; et al. Genomic study of severe fetal anomalies and discovery of GREB1L mutations in renal agenesis. Genet. Med. 2018, 20, 745–753. [Google Scholar] [CrossRef]
- Fu, F.; Li, R.; Li, Y.; Nie, Z.Q.; Lei, T.; Wang, D.; Yang, X.; Han, J.; Pan, M.; Zhen, L.; et al. Whole exome sequencing as a diagnostic adjunct to clinical testing in fetuses with structural abnormalities. Ultrasound Obs. Gynecol. 2018, 51, 493–502. [Google Scholar] [CrossRef]
- Leung, G.K.C.; Mak, C.C.Y.; Fung, J.L.F.; Wong, W.H.S.; Tsang, M.H.Y.; Yu, M.H.C.; Pei, S.L.C.; Yeung, K.S.; Mok, G.T.K.; Lee, C.P.; et al. Identifying the genetic causes for prenatally diagnosed structural congenital anomalies (SCAs) by whole-exome sequencing (WES). BMC Med. Genom. 2018, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Normand, E.A.; Braxton, A.; Nassef, S.; Ward, P.A.; Vetrini, F.; He, W.; Patel, V.; Qu, C.; Westerfield, L.E.; Stover, S.; et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Meier, N.; Bruder, E.; Lapaire, O.; Hoesli, I.; Kang, A.; Hench, J.; Hoeller, S.; De Geyter, J.; Miny, P.; Heinimann, K.; et al. Exome sequencing of fetal anomaly syndromes: Novel phenotype-genotype discoveries. Eur. J. Hum. Genet. 2019, 27, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Daum, H.; Meiner, V.; Elpeleg, O.; Harel, T.; Collaborating Authors. Fetal exome sequencing: Yield and limitations in a tertiary referral center. Ultrasound Obs. Gynecol. 2019, 53, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Quinlan-Jones, E.; Lord, J.; Williams, D.; Hamilton, S.; Marton, T.; Eberhardt, R.Y.; Rinck, G.; Prigmore, E.; Keelagher, R.; McMullan, D.J.; et al. Molecular autopsy by trio exome sequencing (ES) and postmortem examination in fetuses and neonates with prenatally identified structural anomalies. Genet. Med. 2019, 21, 1065–1073. [Google Scholar] [CrossRef]
- De Koning, M.A.; Haak, M.C.; Adama van Scheltema, P.N.; Peeters-Scholte, C.M.P.C.D.; Koopmann, T.T.; Nibbeling, E.A.R.; Aten, E.; den Hollander, N.S.; Ruivenkamp, C.A.L.; Hoffer, M.J.V.; et al. From diagnostic yield to clinical impact: A pilot study on the implementation of prenatal exome sequencing in routine care. Genet. Med. 2019, 21, 2303–2310. [Google Scholar] [CrossRef]
- Lord, J.; McMullan, D.J.; Eberhardt, R.Y.; Rinck, G.; Hamilton, S.J.; Quinlan-Jones, E.; Prigmore, E.; Keelagher, R.; Best, S.K.; Carey, G.K.; et al. Prenatal Assessment of Genomes and Exomes Consortium. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 2019, 393, 747–757. [Google Scholar] [CrossRef]
- Petrovski, S.; Aggarwal, V.; Giordano, J.L.; Stosic, M.; Wou, K.; Bier, L.; Spiegel, E.; Brennan, K.; Stong, N.; Jobanputra, V.; et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: A prospective cohort study. Lancet 2019, 393, 758–767. [Google Scholar] [CrossRef]
- Becher, N.; Andreasen, L.; Sandager, P.; Lou, S.; Petersen, O.B.; Christensen, R.; Vogel, I. Implementation of exome sequencing in fetal diagnostics-Data and experiences from a tertiary center in Denmark. Acta Obs. Gynecol. Scand. 2020, 99, 783–790. [Google Scholar] [CrossRef]
- Chen, M.; Chen, J.; Wang, C.; Chen, F.; Xie, Y.; Li, Y.; Li, N.; Wang, J.; Zhang, V.W.; Chen, D. Clinical application of medical exome sequencing for prenatal diagnosis of fetal structural anomalies. Eur. J. Obs. Gynecol. Reprod. Biol. 2020, 251, 119–124. [Google Scholar] [CrossRef]
- Dempsey, E.; Haworth, A.; Ive, L.; Dubis, R.; Savage, H.; Serra, E.; Kenny, J.; Elmslie, F.; Greco, E.; Thilaganathan, B.; et al. A report on the impact of rapid prenatal exome sequencing on the clinical management of 52 ongoing pregnancies; a retrospective review. BJOG 2020. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Jiang, Y.; Zhou, X.; Meng, H.; Hao, N.; Chang, J.; Bai, J.; Wang, C.; Wang, M.; Guo, J. Simultaneous Detection of CNVs and SNVs Improves the Diagnostic Yield of Fetuses with Ultrasound Anomalies and Normal Karyotypes. Genes 2020, 11, 1397. [Google Scholar] [CrossRef] [PubMed]
- Weitensteiner, V.; Zhang, R.; Bungenberg, J.; Marks, M.; Gehlen, J.; Ralser, D.J.; Hilger, A.C.; Sharma, A.; Schumacher, J.; Gembruch, U.; et al. Exome sequencing in syndromic brain malformations identifies novel mutations in ACTB, and SLC9A6, and suggests BAZ1A as a new candidate gene. Birth. Defects Res. 2018, 110, 587–597. [Google Scholar] [CrossRef]
- Westphal, D.S.; Leszinski, G.S.; Rieger-Fackeldey, E.; Graf, E.; Weirich, G.; Meitinger, T.; Ostermayer, E.; Oberhoffer, R.; Wagner, M. Lessons from exome sequencing in prenatally diagnosed heart defects: A basis for prenatal testing. Clin. Genet. 2019, 95, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shen, M.; Yan, Y.; Tan, Y.; Zhang, J.; Wu, J.; Yang, G.; Li, S.; Wang, J.; Ren, Z.; et al. Genetic Analysis in Fetal Skeletal Dysplasias by Trio Whole-Exome Sequencing. Biomed. Res. Int. 2019, 2019, 2492590. [Google Scholar] [CrossRef]
- Sun, H.; Yi, T.; Hao, X.; Yan, H.; Wang, J.; Li, Q.; Gu, X.; Zhou, X.; Wang, S.; Wang, X.; et al. Contribution of single−gene defects to congenital cardiac left−sided lesions in the prenatal setting. Ultrasound Obs. Gyn. 2020, 56, 225–232. [Google Scholar] [CrossRef]
- Heide, S.; Spentchian, M.; Valence, S.; Buratti, J.; Mach, C.; Lejeune, E.; Olin, V.; Massimello, M.; Lehalle, D.; Mouthon, L.; et al. Prenatal exome sequencing in 65 fetuses with abnormality of the corpus callosum: Contribution to further diagnostic delineation. Genet. Med. 2020, 22, 1887–1891. [Google Scholar] [CrossRef]
- Lei, T.Y.; Fu, F.; Li, R.; Yu, Q.X.; Du, K.; Zhang, W.W.; Deng, Q.; Li, L.S.; Wang, D.; Yang, X.; et al. Whole-exome sequencing in the evaluation of fetal congenital anomalies of the kidney and urinary tract detected by ultrasonography. Prenat. Diagn. 2020, 40, 1290–1299. [Google Scholar] [CrossRef]
- Li, R.; Fu, F.; Yu, Q.; Wang, D.; Jing, X.; Zhang, Y.; Li, F.; Li, F.; Han, J.; Pan, M.; et al. Prenatal exome sequencing in fetuses with congenital heart defects. Clin. Genet. 2020, 98, 215–230. [Google Scholar] [CrossRef]
- Mone, F.; Eberhardt, R.Y.; Morris, R.K.; Hurles, M.E.; McMullan, D.J.; Maher, E.R.; Lord, J.; Chitty, L.S.; Giordano, J.L.; Wapner, R.J.; et al. CODE Study Collaborators. COngenital heart disease and the Diagnostic yield with Exome sequencing (CODE) study: Prospective cohort study and systematic review. Ultrasound Obs. Gynecol. 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Qiao, F.; Wang, Y.; Zhang, C.; Zhou, R.; Wu, Y.; Wang, C.; Meng, L.; Mao, P.; Cheng, Q.; Luo, C.; et al. Comprehensive evaluation of genetic variants in fetuses with congenital heart defect using chromosomal microarray analysis and exome sequencing. Ultrasound Obs. Gynecol. 2020. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Sparks, T.N.; Lianoglou, B.R.; Adami, R.R.; Pluym, I.D.; Holliman, K.; Duffy, J.; Downum, S.L.; Patel, S.; Faubel, A.; Boe, N.M.; et al. University of California Fetal–Maternal Consortium; University of California, San Francisco Center for Maternal–Fetal Precision Medicine. Exome Sequencing for Prenatal Diagnosis in Nonimmune Hydrops Fetalis. N. Engl. J. Med. 2020, 383, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Xie, Y.; Chen, F.; Chen, M.; Yu, L.; Chen, D.; Chen, J. Novel and recurrent variants identified in fetuses with central nervous system abnormalities by trios-medical exome sequencing. Clin. Chim. Acta 2020, 510, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhou, C.; Shi, H.; Mo, Y.; Tan, W.; Sun, T.; Zhu, J.; Li, Q.; Li, H.; Li, Y.; et al. Prenatal diagnosis of skeletal dysplasias using whole exome sequencing in China. Clin. Chim. Acta 2020, 507, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Yan, H.; Chen, J.; Li, N.; Wang, J.; Liu, Y.; Zhang, H.; Li, S.; Zhang, W.; Chen, D.; et al. Genetic Examination for Fetuses with Increased Fetal Nuchal Translucency by Genomic Technology. Cytogenet. Genome Res. 2020, 60, 57–62. [Google Scholar] [CrossRef]
- Yang, X.; Huang, L.Y.; Pan, M.; Xu, L.L.; Zhen, L.; Han, J.; Li, D.Z. Exome sequencing improves genetic diagnosis of fetal increased nuchal translucency. Prenat. Diagn. 2020, 40, 1426–1431. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Shao, B.; Wang, C.; Hu, P.; Qiao, F.; Xu, Z. Molecular diagnostic in fetuses with isolated congenital anomalies of the kidney and urinary tract by whole-exome sequencing. J. Clin. Lab. Anal. 2020, 34, e23480. [Google Scholar] [CrossRef]
- Han, J.; Yang, Y.D.; He, Y.; Liu, W.J.; Zhen, L.; Pan, M.; Yang, X.; Zhang, V.W.; Liao, C.; Li, D.Z. Rapid prenatal diagnosis of skeletal dysplasia using medical trio exome sequencing: Benefit for prenatal counseling and pregnancy management. Prenat. Diagn. 2020, 40, 577–584. [Google Scholar] [CrossRef]
- Li, L.; Fu, F.; Li, R.; Xiao, W.; Yu, Q.; Wang, D.; Jing, X.; Zhang, Y.; Yang, X.; Pan, M.; et al. Genetic tests aid in counseling of fetuses with cerebellar vermis defects. Prenat. Diagn. 2020, 40, 1228–1238. [Google Scholar] [CrossRef]
- Carss, K.J.; Hillman, S.C.; Parthiban, V.; McMullan, D.J.; Maher, E.R.; Kilby, M.D.; Hurles, M.E. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum. Mol. Genet. 2014, 23, 3269–3277. [Google Scholar] [CrossRef]
- Mackie, F.L.; Carss, K.J.; Hillman, S.C.; Hurles, M.E.; Kilby, M.D. Exome Sequencing in Fetuses with Structural Malformations. J. Clin. Med. 2014, 3, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, L.E.; Stover, S.R.; Mathur, V.S.; Nassef, S.A.; Carter, T.G.; Yang, Y.; Eng, C.M.; Van den Veyver, I.B. Reproductive genetic counseling challenges associated with diagnostic exome sequencing in a large academic private reproductive genetic counseling practice. Prenat. Diagn. 2015, 35, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Alamillo, C.L.; Powis, Z.; Farwell, K.; Shahmirzadi, L.; Weltmer, E.C.; Turocy, J.; Lowe, T.; Kobelka, C.; Chen, E.; Basel, D.; et al. Exome sequencing positively identified relevant alterations in more than half of cases with an indication of prenatal ultrasound anomalies. Prenat. Diagn. 2015, 35, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Kurdi, W.; Almusafri, F.; Alnemer, M.; Alkaff, A.; Babay, Z.; Alhashem, A.; Tulbah, M.; Alsahan, N.; Khan, R.; et al. Molecular autopsy in maternal-fetal medicine. Genet. Med. 2018, 20, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Stals, K.L.; Wakeling, M.; Baptista, J.; Caswell, R.; Parrish, A.; Rankin, J.; Tysoe, C.; Jones, G.; Gunning, A.C.; Lango Allen, H.; et al. Diagnosis of lethal or prenatal-onset autosomal recessive disorders by parental exome sequencing. Prenat. Diagn. 2018, 38, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.; Gilmore, K.; Hardisty, E.; Lyerly, A.D.; Vora, N.L. Ethical and counseling challenges in prenatal exome sequencing. Prenat. Diagn. 2018, 38, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Aarabi, M.; Sniezek, O.; Jiang, H.; Saller, D.N.; Bellissimo, D.; Yatsenko, S.A.; Rajkovic, A. Importance of complete phenotyping in prenatal whole exome sequencing. Hum. Genet. 2018, 137, 175–181. [Google Scholar] [CrossRef]
- Greenbaum, L.; Pode-Shakked, B.; Eisenberg-Barzilai, S.; Dicastro-Keidar, M.; Bar-Ziv, A.; Goldstein, N.; Reznik-Wolf, H.; Poran, H.; Rigbi, A.; Barel, O.; et al. Evaluation of Diagnostic Yield in Fetal Whole-Exome Sequencing: A Report on 45 Consecutive Families. Front. Genet. 2019, 25, 425. [Google Scholar] [CrossRef]
- Aoi, H.; Mizuguchi, T.; Suzuki, T.; Makino, S.; Yamamoto, Y.; Takeda, J.; Maruyama, Y.; Seyama, R.; Takeuchi, S.; Uchiyama, Y.; et al. Whole exome sequencing of fetal structural anomalies detected by ultrasonography. J. Hum. Genet. 2020. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Quaio, C.R.D.C.; Moreira, C.M.; Novo-Filho, G.M.; Sacramento-Bobotis, P.R.; Groenner Penna, M.; Perazzio, S.F.; Dutra, A.P.; da Silva, R.A.; Santos, M.N.P.; de Arruda, V.Y.N.; et al. Diagnostic power and clinical impact of exome sequencing in a cohort of 500 patients with rare diseases. Am. J. Med. Genet. C Semin. Med. Genet. 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Vora, N.L.; Gilmore, K.; Brandt, A.; Gustafson, C.; Strande, N.; Ramkissoon, L.; Hardisty, E.; Foreman, A.K.M.; Wilhelmsen, K.; Owen, P.; et al. An approach to integrating exome sequencing for fetal structural anomalies into clinical practice. Genet. Med. 2020, 22, 954–961. [Google Scholar] [CrossRef] [PubMed]
- De Tomasi, L.; David, P.; Humbert, C.; Silbermann, F.; Arrondel, C.; Tores, F.; Fouquet, S.; Desgrange, A.; Niel, O.; Bole-Feysot, C.; et al. Mutations in GREB1L Cause Bilateral Kidney Agenesis in Humans and Mice. Am. J. Hum. Genet. 2017, 101, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.Y.; Fu, F.; Li, R.; Wang, D.; Wang, R.Y.; Jing, X.Y.; Deng, Q.; Li, Z.Z.; Liu, Z.Q.; Yang, X.; et al. Whole-exome sequencing for prenatal diagnosis of fetuses with congenital anomalies of the kidney and urinary tract. Nephrol. Dial. Transpl. 2017, 32, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Sunde, L.; Nielsen, M.L.; Ramsing, M.; Petersen, A.; Hjortshøj, T.D.; Olsen, T.E.; Tabor, A.; Hertz, J.M.; Johnsen, I.; et al. Targeted gene sequencing and whole-exome sequencing in autopsied fetuses with prenatally diagnosed kidney anomalies. Clin. Genet. 2018, 93, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Reches, A.; Hiersch, L.; Simchoni, S.; Barel, D.; Greenberg, R.; Ben Sira, L.; Malinger, G.; Yaron, Y. Whole-exome sequencing in fetuses with central nervous system abnormalities. J. Perinatol. 2018, 38, 1301–1308. [Google Scholar] [CrossRef]
- Mary, L.; Chennen, K.; Stoetzel, C.; Antin, M.; Leuvrey, A.; Nourisson, E.; Alanio-Detton, E.; Antal, M.C.; Attié-Bitach, T.; Bouvagnet, P.; et al. Bardet-Biedl syndrome: Antenatal presentation of forty-five fetuses with biallelic pathogenic variants in known Bardet-Biedl syndrome genes. Clin. Genet. 2019, 95, 384–397. [Google Scholar] [CrossRef]
- Chandler, N.; Best, S.; Hayward, J.; Faravelli, F.; Mansour, S.; Kivuva, E.; Tapon, D.; Male, A.; DeVile, C.; Chitty, L.S. Rapid prenatal diagnosis using targeted exome sequencing: A cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet. Med. 2018, 20, 1430–1437. [Google Scholar] [CrossRef]
- Yauy, K.; Tran Mau-Them, F.; Willems, M.; Coubes, C.; Blanchet, P.; Herlin, C.; Taleb Arrada, I.; Sanchez, E.; Faure, J.M.; Le Gac, M.P.; et al. B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet. Med. 2018, 20, 269–274. [Google Scholar] [CrossRef]
- Van Nisselrooij, A.E.L.; Lugthart, M.A.; Clur, S.A.; Linskens, I.H.; Pajkrt, E.; Rammeloo, L.A.; Rozendaal, L.; Blom, N.A.; van Lith, J.M.M.; Knegt, A.C.; et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet. Med. 2020, 22, 1206–1214. [Google Scholar] [CrossRef]
- Talkowski, M.E.; Ordulu, Z.; Pillalamarri, V.; Benson, C.B.; Blumenthal, I.; Connolly, S.; Hanscom, C.; Hussain, N.; Pereira, S.; Picker, J.; et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N. Engl. J. Med. 2012, 367, 2226–2232. [Google Scholar] [CrossRef]
- Filges, I.; Friedman, J.M. Exome sequencing for gene discovery in lethal fetal disorders—Harnessing the value of extreme phenotypes. Prenat. Diagn. 2015, 35, 1005–1009. [Google Scholar] [CrossRef]
- Hillman, S.C.; Willams, D.; Carss, K.J.; McMullan, D.J.; Hurles, M.E.; Kilby, M.D. Prenatal exome sequencing for fetuses with structural abnormalities: The next step. Ultrasound Obs. Gynecol. 2015, 45, 4–9. [Google Scholar] [CrossRef]
- International Society for Prenatal Diagnosis; Society for Maternal and Fetal Medicine; Perinatal Quality Foundation. Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat. Diagn. 2018, 38, 6–9. [Google Scholar] [CrossRef]
- Monaghan, K.G.; Leach, N.T.; Pekarek, D.; Prasad, P.; Rose, N.C.; ACMG Professional Practice and Guidelines Committee. The use of fetal exome sequencing in prenatal diagnosis: A points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 675–680. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Mellis, R.; Chandler, N.; Chitty, L.S. Next-generation sequencing and the impact on prenatal diagnosis. Expert Rev. Mol. Diagn. 2018, 18, 689–699. [Google Scholar] [CrossRef]
- Ferretti, L.; Mellis, R.; Chitty, L.S. Update on the use of exome sequencing in the diagnosis of fetal abnormalities. Eur. J. Med. Genet. 2019, 62, 103663. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Spinner, N.B.; Rehm, H.L.; Green, R.C.; Bianchi, D.W. Prenatal DNA Sequencing: Clinical, Counseling, and Diagnostic Laboratory Considerations. Prenat. Diagn. 2018, 38, 26–32. [Google Scholar] [CrossRef]
- Ewans, L.J.; Schofield, D.; Shrestha, R.; Zhu, Y.; Gayevskiy, V.; Ying, K.; Walsh, C.; Lee, E.; Kirk, E.P.; Colley, A.; et al. Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet. Med. 2018, 20, 1564–1574. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef]
- ACMG Board of Directors. ACMG policy statement: Updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet. Med. 2015, 17, 68–69. [Google Scholar] [CrossRef]
- Brew, C.E.; Castro, B.A.; Pan, V.; Hart, A.; Blumberg, B.; Wicklund, C. Genetics professionals’ attitudes toward prenatal exome sequencing. J. Genet. Couns. 2019, 28, 229–239. [Google Scholar] [CrossRef]
- Narayanan, S.; Blumberg, B.; Clayman, M.L.; Pan, V.; Wicklund, C. Exploring the Issues Surrounding Clinical Exome Sequencing in the Prenatal Setting. J. Genet. Couns. 2018, 27, 1228–1237. [Google Scholar]
- Richardson, A.; Ormond, K.E. Ethical considerations in prenatal testing: Genomic testing and medical uncertainty. Semin. Fetal Neonatal Med. 2018, 23, 1–6. [Google Scholar] [CrossRef]
- Quinlan-Jones, E.; Kilby, M.D.; Greenfield, S.; Parker, M.; McMullan, D.; Hurles, M.E.; Hillman, S.C. Prenatal whole exome sequencing: The views of clinicians, scientists, genetic counsellors and patient representatives. Prenat. Diagn. 2016, 36, 935–941. [Google Scholar] [CrossRef]
- Bunnik, E.M.; de Jong, A.; Nijsingh, N.; de Wert, G.M. The new genetics and informed consent: Differentiating choice to preserve autonomy. Bioethics 2013, 27, 348–355. [Google Scholar] [CrossRef]
- Best, S.; Wou, K.; Vora, N.; Van der Veyver, I.B.; Wapner, R.; Chitty, L.S. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat. Diagn. 2018, 38, 10–19. [Google Scholar] [CrossRef]
- Werner-Lin, A.; Mccoyd, J.L.M.; Bernhardt, B.A. Actions and Uncertainty: How Prenatally Diagnosed Variants of Uncertain Significance Become Actionable. Hastings Cent. Rep. 2019, 49 (Suppl. 1), S61–S71. [Google Scholar] [CrossRef]
- The Lancet. Fetal medicine: Past, present, and future. Lancet 2019, 393, 717. [Google Scholar] [CrossRef]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef]
- Chitty, L.S. Advances in the prenatal diagnosis of monogenic disorders. Prenat. Diagn. 2018, 38, 3–5. [Google Scholar] [CrossRef]
- Vora, N.L.; Hui, L. Next-generation sequencing and prenatal ’omics: Advanced diagnostics and new insights into human development. Genet. Med. 2018, 20, 791–799. [Google Scholar] [CrossRef]
- Wise, A.L.; Manolio, T.A.; Mensah, G.A.; Peterson, J.F.; Roden, D.M.; Tamburro, C.; Williams, M.S.; Green, E.D. Genomic medicine for undiagnosed diseases. Lancet 2019, 394, 533–540. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2020, 49, 1207–1217. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Article | Test | Geographic Area | Diagnostic Yield (%) | Inconclusive Findings (%) | VUS (%) | Novel Candidate Genes (%) | Negative (%) | Secondary/ Incidental Findings |
|---|---|---|---|---|---|---|---|---|
| Pangalos, 2016 [26] | in silico panel from WES data | Greece | 6/14 (43%) | 1/14 (7%) | not provided | not provided | 7/14 (50%) | not provided |
| Vora, 2017 [27] | in silico panel from WES data | USA | 7/15 (47%) | 3/15 (20%) | 2/15 (13%) | 1/15 (7%) | 5/15 (33%) | not provided |
| Yates, 2017 [28] | WES | USA | 17/84 (20%) | 45/84 (54%) | 38/84 (45%) | 7/84 (9%) | 22/84 (26%) | 2 |
| Boissel, 2018 [29] | WES | Canada | 19/101 (19%) | 5/101 (5%) | 1/101 (1%) | 4/101 (4%) | 77/101 (76%) | not provided |
| Fu, 2018 [30] | WES | China | 47/196 (24%) | 25/196 (13%) | 25/196 (13%) | 0 | 124/196 (63%) | 12 |
| Leung, 2018 [31] | WES | China | 3/33 (9%) | 6/33 (18%) | 6/33 (18%) | 0 | 24/33 (73%) | not provided |
| Normand, 2018 [32] | WES | USA | 46/133 (35%) | not provided | not provided | not provided | --- | not provided |
| Meier, 2019 [33] | WES | Switzerland | 11/26 (42%) | 3/26 (12%) | 0 | 3/26 (12%) | 12/26 (46%) | 3 |
| Daum, 2019 [34] | WES | Israel | 16/77 (21%) | not provided | not provided | not provided | --- | not provided |
| Quinlan-Jones, 2019 [35] | in silico panel from WES data | UK | 10/25 (40%) | 6/25 (24%) | 6/25 (24%) | 0 | 9/25 (36%) | not provided |
| De Koning, 2019 [36] | WES | Netherlands | 8/20 (40%) | 0/20 (0%) | 0 | 0 | 12/20 (60%) | 3 |
| Lord, 2019 [37] | WES | UK | 52/610 (9%) | 24/610 (4%) | 24/610 (4%) | 0 | 534/610 (88%) | not provided |
| Petrovski, 2019 [38] | WES | USA | 24/234 (10%) | 46/234 (20%) | not provided | not provided | 164/234 (70%) | 4 |
| Becher, 2020 [39] | WES | Denmark | 9/35 (26%) | 7/35 (20%) | 7/35 (20%) | 0 | 19/35 (54%) | 1 |
| Chen, 2020 [40] | CES | China | 20/105 (19%) | 12/105 (11%) | 12/105 (11%) | 0 | 73/105 (70%) | not provided |
| Dempsey, 2020 [41] | CES | UK | 18/52 (35%) | 13/52 (25%) | 13/52 (25%) | 0 | 21/52 (40%) | not provided |
| Qi, 2020 [42] | CES | China | 27/80 (34%) | 5/80 (6%) | 5/80 (6%) | 0 | 48/80 (60%) | not provided |
| Article | Anomaly | Test | Geographic Area | Diagnostic Yield (%) | VUS (%) | Novel Candidate Genes (%) | Negative (%) | Secondary/ Incidental Findings |
|---|---|---|---|---|---|---|---|---|
| Weitensteiner, 2018 [43] | Brain malformations | WES | Germany | 3/6 (50%) | 0 | 0 | 3/6 (50%) | 1 |
| Westphal, 2019 [44] | Congenital heart diseases | CES | Germany | 6/30 (20%) | 2/30 (7%) | 2/30 (7%) | 20/30 (67%) | 3 |
| Yang, 2019 [45] | Skeletal dysplasias | CES | China | 6/8 (75%) | 0 | 0 | 2/8 (25%) | not provided |
| Sun, 2020 [46] | Congenital cardiac left-sided lesions | WES | China | 13/66 (20%) | 5/66 (8%) | 6/66 (9%) | 42/66 (64%) | 1 |
| Heide, 2020 [47] | Corpus callosum abnormalities | WES | France | 12/62 (19%) | 6/62 (10%) | 0 | 44/62 (71%) | not provided |
| Lei, 2020 [48] | Congenital anomalies of the kidney and urinary tract | WES | China | 12/163 (12%) | 2/163 (1%) | 0 | 149/163 (91%) | 9 |
| Li R., 2020 [49] | Congenital heart disease | WES | China | 26/260 (10%) | 16/260 (6%) | 16/260 (6%) | 202/260 (78%) | 7 |
| Mone, 2020 [50] | Congenital heart disease | in silico panel from WES data | UK | 25/197 (13%) | 10/197 (5%) | 0 | 162/197 (82%) | not provided |
| Qiao, 2020 [51] | Congenital heart disease | WES | China | 24/300 (8%) | 32/300 (11%) | 0 | 244/300 (81%) | 48 |
| Sparks, 2020 [52] | Non-immune hydrops fetalis | WES | USA | 37/127 (29%) | 12/127 (9%) | 0 | 78/127 (61.41%) | 4 |
| Tan, 2020 [53] | Brain anomalies | CES | China | 5/11 (45%) | 0 | 0 | 6/11 (55%) | not provided |
| Tang, 2020 [54] | Skeletal dysplasias | WES | China | 6/8 (75%) | 0 | 0 | 2/8 (25%) | not provided |
| Xue, 2020 [55] | Increased nuchal translucency | WES | China | 3/24 (13%) | 0 | 0 | 21/24 (88%) | 2 |
| Yang, 2020 [56] | Increased nuchal translucency | CES | China | 4/73 (6%) | 0 | 0 | 69/73 (95%) | 7 |
| Zhou, 2020 [57] | Congenital anomalies of the kidney and urinary tract | WES | China | 3/41 (7%) | 0 | 0 | 1 | |
| Han, 2020 [58] | Skeletal dysplasias | CES | China | 24/26 (92%) | 0 | 0 | 2/26 (8%) | not provided |
| Li L., 2020 [59] | Cerebellar vermis defects | WES | China | 8/19 (42%) | 2/19 (11%) | 0 | 9/19 (47%) | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guadagnolo, D.; Mastromoro, G.; Di Palma, F.; Pizzuti, A.; Marchionni, E. Prenatal Exome Sequencing: Background, Current Practice and Future Perspectives—A Systematic Review. Diagnostics 2021, 11, 224. https://doi.org/10.3390/diagnostics11020224
Guadagnolo D, Mastromoro G, Di Palma F, Pizzuti A, Marchionni E. Prenatal Exome Sequencing: Background, Current Practice and Future Perspectives—A Systematic Review. Diagnostics. 2021; 11(2):224. https://doi.org/10.3390/diagnostics11020224
Chicago/Turabian StyleGuadagnolo, Daniele, Gioia Mastromoro, Francesca Di Palma, Antonio Pizzuti, and Enrica Marchionni. 2021. "Prenatal Exome Sequencing: Background, Current Practice and Future Perspectives—A Systematic Review" Diagnostics 11, no. 2: 224. https://doi.org/10.3390/diagnostics11020224
APA StyleGuadagnolo, D., Mastromoro, G., Di Palma, F., Pizzuti, A., & Marchionni, E. (2021). Prenatal Exome Sequencing: Background, Current Practice and Future Perspectives—A Systematic Review. Diagnostics, 11(2), 224. https://doi.org/10.3390/diagnostics11020224