
Whole-Genome Sequencing for Investigating a Health Care-Associated Outbreak of Carbapenem-Resistant Acinetobacter baumannii

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Whole-Genome Sequencing
2.1.1. Multilocus Sequence Typing
2.1.2. Single Nucleotide Variation Analysis
2.1.3. Cluster Analysis
2.1.4. Antimicrobial Resistance Prediction
2.2. Repetitive Element PCR
3. Results
3.1. Whole-Genome Sequencing Results
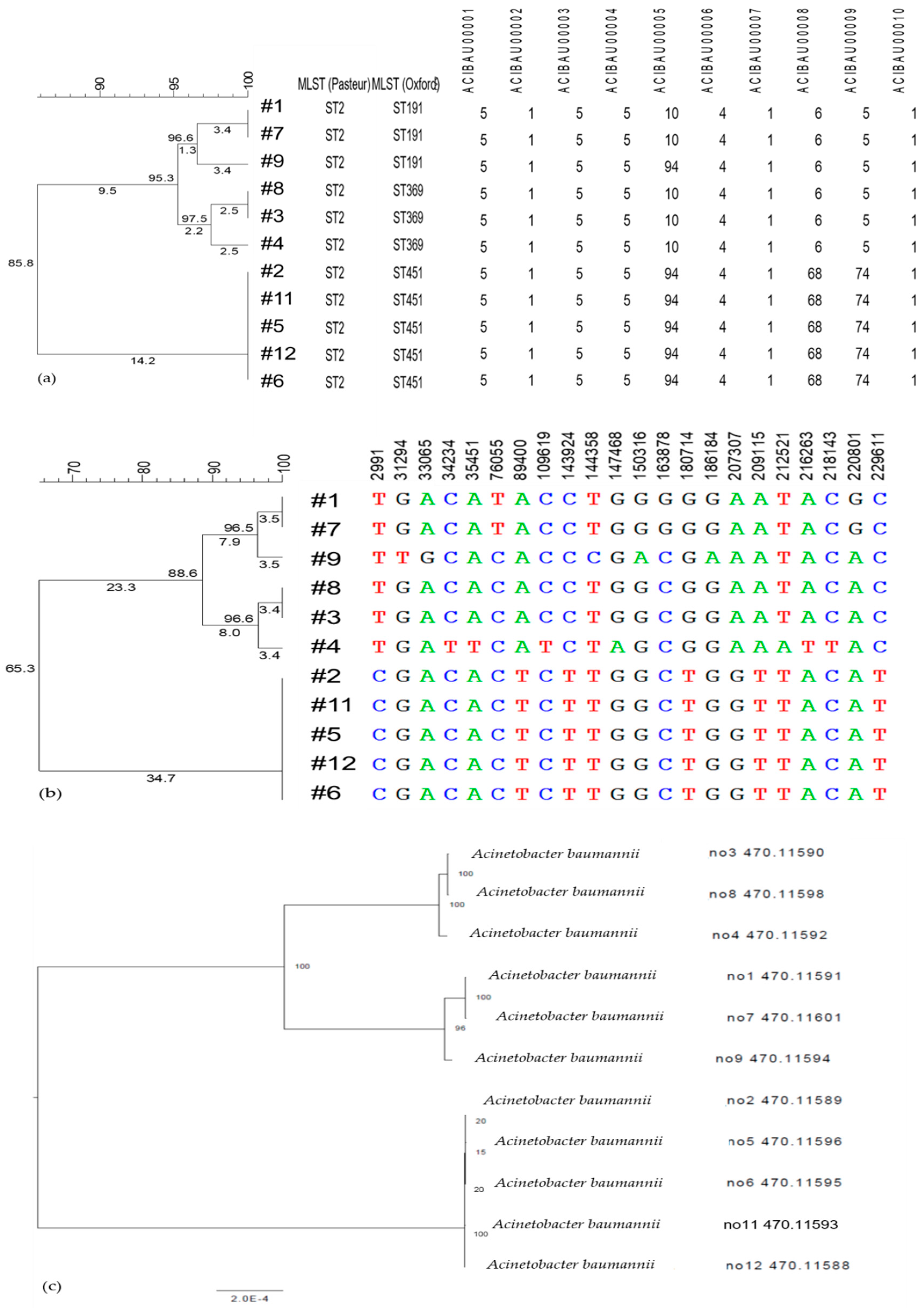
3.1.1. MLST
3.1.2. Single Nucleotide (SNV/SNP) Analysis
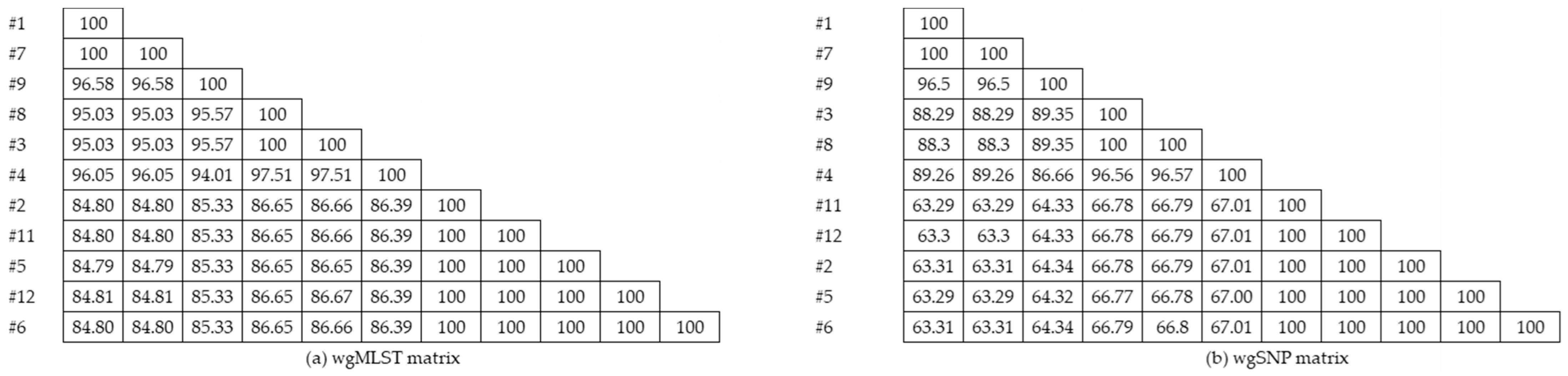
3.1.3. Cluster Analysis
3.1.4. Antimicrobial Resistance Prediction
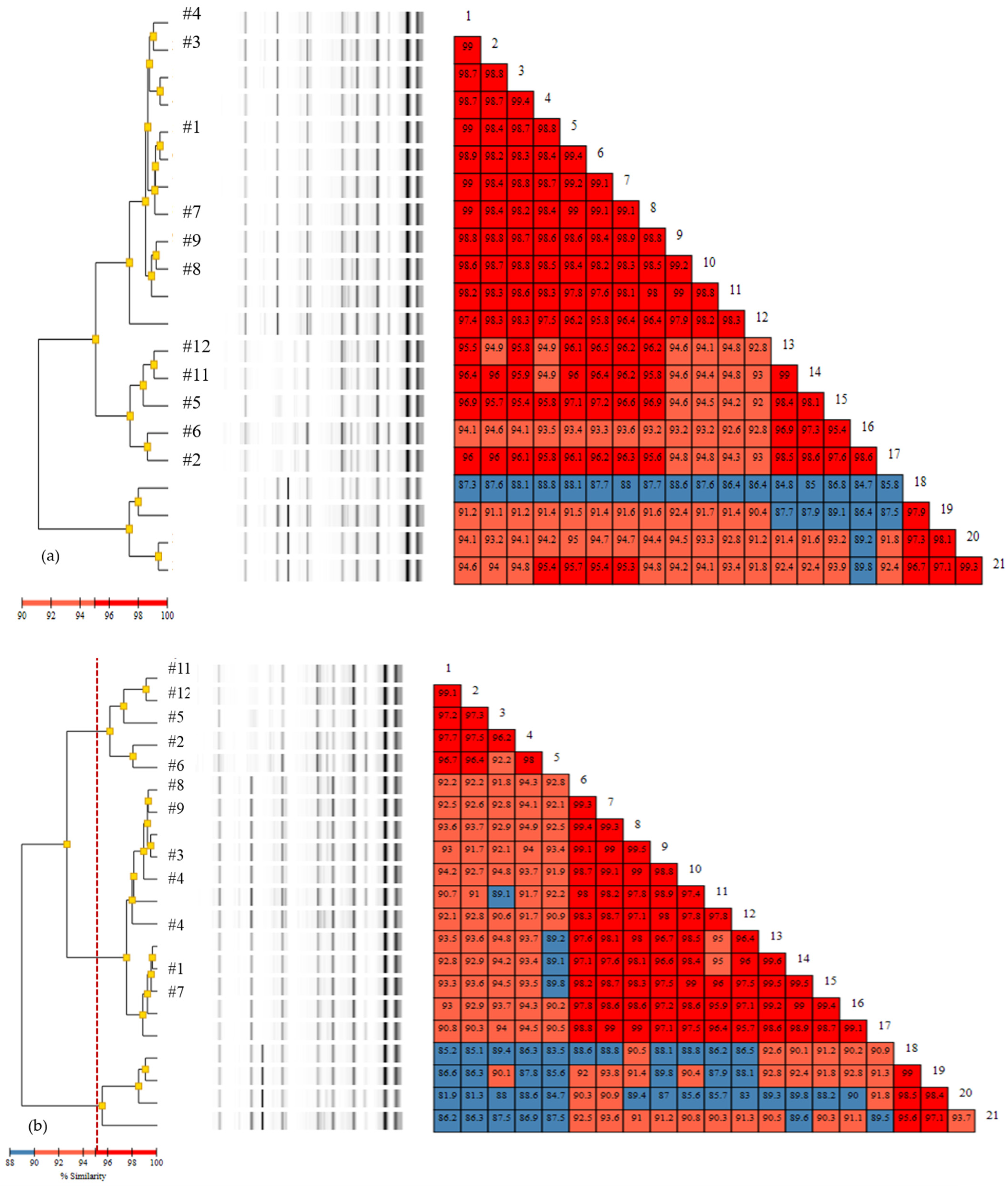
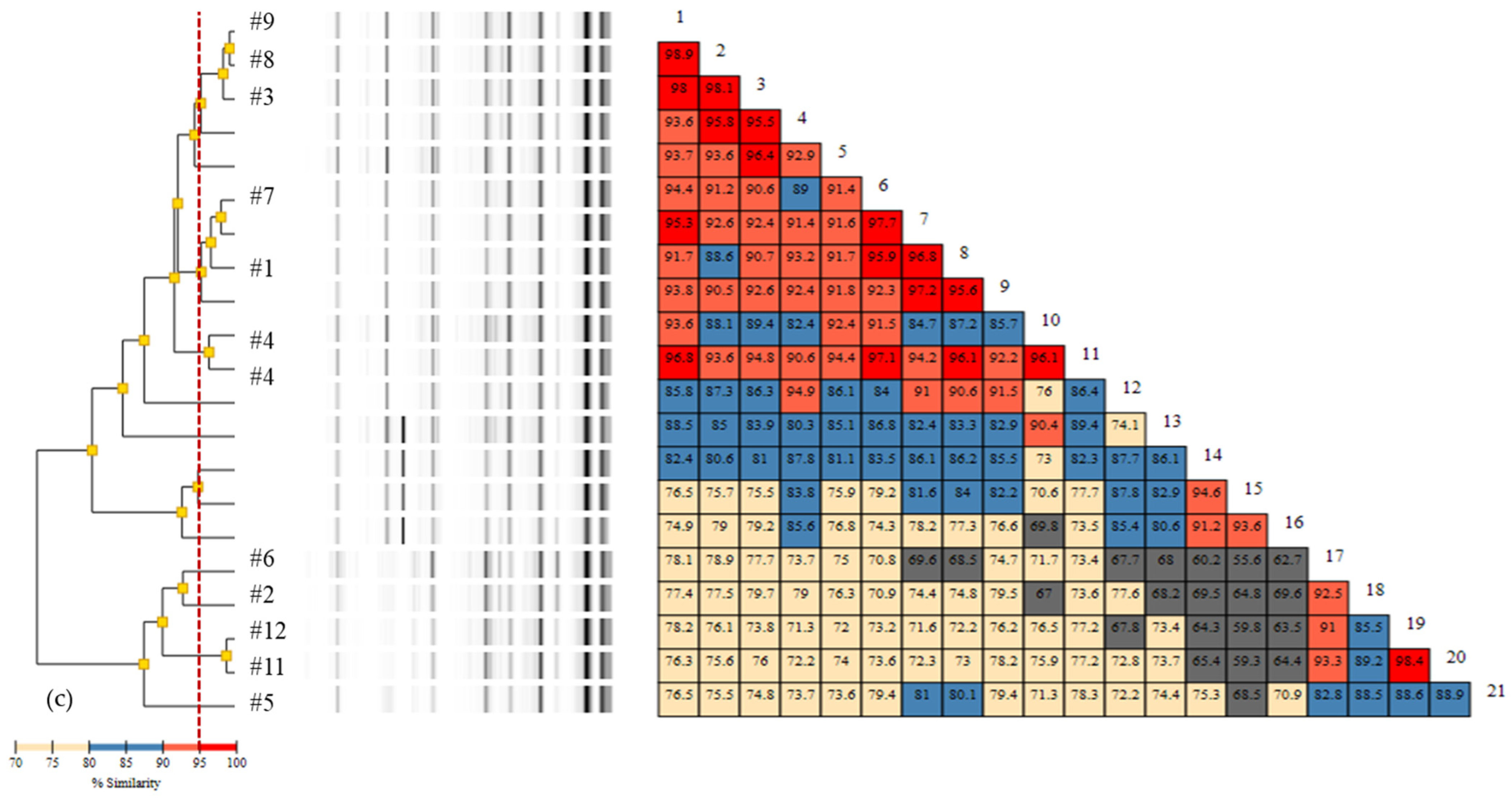
3.2. Rep-PCR
3.3. Comparison of Typing Methods
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dijkshoorn, L.; Nemec, A.; Seifert, H. An increasing threat in hospitals: Multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 2007, 5, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Webb, A.K.; Limbago, B.; Dudeck, M.A.; Patel, J.; Kallen, A.J.; Edwards, J.R.; Sievert, D.M. Antimicrobial-resistant pathogens associated with healthcare-associated infections: Summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect. Control. Hosp. Epidemiol. 2016, 37, 1288–1301. [Google Scholar] [CrossRef] [PubMed]
- Wendt, C.; Dietze, B.; Dietz, E.; Rüden, H. Survival of Acinetobacter baumannii on dry surfaces. J. Clin. Microbiol. 1997, 35, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Magrini, N.; Kahlmeter, G.; Singh, N.J. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Isler, B.; Doi, Y.; Bonomo, R.A.; Paterson, D.L. New treatment options against carbapenem-resistant Acinetobacter baumannii infections. Antimicrob. Agents Chemother. 2018, 63, e01110-18. [Google Scholar] [CrossRef]
- Kalenić, S.; Budimir, A.J. The role of the microbiology laboratory in healthcare-associated infection prevention. Int. J. Infect. Control. 2009, 5. [Google Scholar] [CrossRef]
- Tenover, F.C.; Arbeit, R.D.; Goering, R.V. How to select and interpret molecular strain typing methods for epidemiological studies of bacterial infections a review for healthcare epidemiologists. Infect. Control. Hosp. Epidemiol. 1997, 18, 426–439. [Google Scholar] [CrossRef]
- MacCannell, D. Bacterial strain typing. Clin. Lab. Med. 2013, 33, 629–650. [Google Scholar] [CrossRef]
- Li, W.; Raoult, D.; Fournier, P.E. Bacterial strain typing in the genomic era. FEMS Microbiol. Rev. 2009, 33, 892–916. [Google Scholar] [CrossRef]
- Quainoo, S.; Coolen, J.P.; van Hijum, S.A.; Huynen, M.A.; Melchers, W.J.; van Schaik, W.; Wertheim, H.F. Whole-genome sequencing of bacterial pathogens: The future of nosocomial outbreak analysis. Clin. Microbiol. Rev. 2017, 30, 1015–1063. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A.; Hewlett, E.L. Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef]
- Harris, S.R.; Cartwright, E.J.; Török, M.E.; Holden, M.T.; Brown, N.M.; Ogilvy-Stuart, A.L.; Ellington, M.J.; Quail, M.A.; Bentley, S.D.; Parkhill, J.J. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: A descriptive study. Lancet Infect. Dis. 2013, 13, 130–136. [Google Scholar] [CrossRef]
- Durand, G.; Javerliat, F.; Bes, M.; Veyrieras, J.-B.; Guigon, G.; Mugnier, N.; Schicklin, S.; Kaneko, G.; Santiago-Allexant, E. Bouchiat Routine whole-genome sequencing for outbreak investigations of Staphylococcus aureus in a national reference center. Front. Microbiol. 2018, 9, 511. [Google Scholar] [CrossRef] [PubMed]
- Köser, C.U.; Ellington, M.J.; Cartwright, E.J.; Gillespie, S.H.; Brown, N.M.; Farrington, M.; Holden, M.T.; Dougan, G.; Bentley, S.D.; Parkhill, J.J. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012, 8, e1002824. [Google Scholar] [CrossRef] [PubMed]
- Rossen, J.W.; Friedrich, A.; Moran-Gilad, J.J. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect. 2018, 24, 355–360. [Google Scholar] [CrossRef]
- Bogaerts, B.; Winand, R.; Fu, Q.; Van Braekel, J.; Ceyssens, P.-J.; Mattheus, W.; Bertrand, S.; De Keersmaecker, S.C.; Roosens, N.H.; Vanneste, K.J. Validation of a bioinformatics workflow for routine analysis of whole-genome sequencing data and related challenges for pathogen typing in a European National Reference Center: Neisseria meningitidis as a proof-of-concept. Front. Microbiol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Fricke, W.F.; Rasko, D.A. Bacterial genome sequencing in the clinic: Bioinformatic challenges and solutions. Nat. Rev. Genet. 2014, 15, 49–55. [Google Scholar] [CrossRef]
- Neher, R.A.; Bedford, T.J. Real-time analysis and visualization of pathogen sequence data. J. Clin. Microbiol. 2018, 56, e00480-18. [Google Scholar] [CrossRef]
- Snyder, E.; Kampanya, N.; Lu, J.; Nordberg, E.K.; Karur, H.; Shukla, M.; Soneja, J.; Tian, Y.; Xue, T.; Yoo, H.J. PATRIC: The VBI pathosystems resource integration center. Nucleic Acids Res. 2007, 35 (Suppl. S1), D401–D406. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Approved Standard M100-S30; 30th Informational Supplement; Clinical Laboratory Standards Institute: Wayne, PA, USA, 2020. [Google Scholar]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.J. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Bartual, S.G.; Seifert, H.; Hippler, C.; Luzon, M.A.; Wisplinghoff, H.; Rodríguez-Valera, F.J. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J. Clin. Microbiol. 2005, 43, 4382–4390. [Google Scholar] [CrossRef] [PubMed]
- Diancourt, L.; Passet, V.; Nemec, A.; Dijkshoorn, L.; Brisse, S. The population structure of Acinetobacter baumannii: Expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS ONE 2010, 5, e10034. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.-R.; Florensa, A.F. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.T.; Zankari, E.; Aarestrup, F.M.; Lund, O. Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J. Antimicrob. Chemother. 2016, 71, 2484–2488. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.T.; Aarestrup, F.M.; Lund, O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef]
- Castillo-Ramírez, S.; Graña-Miraglia, L.J. Inaccurate multilocus sequence typing of Acinetobacter baumannii. Emerg. Infect. Dis. 2019, 25, 186–187. [Google Scholar] [CrossRef]
- Hua, X.; Zhang, L.; He, J.; Leptihn, S.; Yu, Y.J. Population Biology and Epidemiological Studies of Acinetobacter baumannii in the Era of Whole Genome Sequencing: Is the Oxford Scheme Still Appropriate? Front. Microbiol. 2020, 11, 775. [Google Scholar] [CrossRef]
- Graña-Miraglia, L.; Lozano, L.F.; Velázquez, C.; Volkow-Fernández, P.; Pérez-Oseguera, Á.; Cevallos, M.A.; Castillo-Ramírez, S.J. Rapid gene turnover as a significant source of genetic variation in a recently seeded population of a healthcare-associated pathogen. Front. Microbiol. 2017, 8, 1817. [Google Scholar] [CrossRef]
- Snitkin, E.S.; Zelazny, A.M.; Montero, C.I.; Stock, F.; Mijares, L.; Murray, P.R.; Segre, J.A. Genome-wide recombination drives diversification of epidemic strains of Acinetobacter baumannii. Proc. Natl. Acad. Sci. USA 2011, 108, 13758–13763. [Google Scholar] [CrossRef]
- Lee, S.Y.; Oh, M.H.; Yun, S.H.; Choi, C.W.; Park, E.C.; Song, H.S.; Lee, H.; Yi, Y.S.; Shin, J.; Chung, C.J. Genomic characterization of extensively drug-resistant Acinetobacter baumannii strain; KAB03 belonging to ST451 from Korea. Infect. Genet. Evol. 2018, 65, 150–158. [Google Scholar] [CrossRef]
- Yoon, E.J.; Kim, D.; Lee, H.; Lee, H.S.; Shin, J.H.; Uh, Y.; Shin, K.S.; Kim, Y.A.; Park, Y.S.; Shin, J.H. Counter clinical prognoses of patients with bloodstream infections between causative Acinetobacter baumannii clones ST191 and ST451 belonging to the international clonal lineage II. Front. Public. Health 2019, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, M.A.; Ozer, E.A.; Hauser, A.R. Utility of whole-genome sequencing in characterizing Acinetobacter epidemiology and analyzing hospital outbreaks. J. Clin. Microbiol. 2016, 54, 593–612. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L. Improvements to PATRIC; the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Dammhayn, C.; Hackel, M.; Seifert, H.J. Global spread of carbapenem-resistant Acinetobacter baumannii. J. Antimicrob. Chemother. 2010, 65, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Venditti, C.; Vulcano, A.; D’Arezzo, S.; Gruber, C.; Selleri, M.; Antonini, M.; Lanini, S.; Marani, A.; Puro, V.; Nisii, C.J. Epidemiological investigation of an Acinetobacter baumannii outbreak using core genome multilocus sequence typing. J. Glob. Antimicrob. Resist. 2019, 17, 245–249. [Google Scholar] [CrossRef]
- Higgins, P.G.; Hujer, A.M.; Hujer, K.M.; Bonomo, R.A.; Seifert, H.J. Interlaboratory reproducibility of DiversiLab rep-PCR typing and clustering of Acinetobacter baumannii isolates. J. Med. Microbiol. 2012, 61 Pt 1, 137–141. [Google Scholar] [CrossRef]
- Rafei, R.; Kempf, M.; Eveillard, M.; Dabboussi, F.; Hamze, M.; Joly-Guillou, M.-L. Current molecular methods in epidemiological typing of Acinetobacter baumannii. Future Microbiol. 2014, 9, 1179–1194. [Google Scholar] [CrossRef]
- Lewis, T.; Loman, N.; Bingle, L.; Jumaa, P.; Weinstock, G.; Mortiboy, D.; Pallen, M.J. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J. Hosp. Infect. 2010, 75, 37–41. [Google Scholar] [CrossRef]
- Goering, R.V.; Köck, R.; Grundmann, H.; Werner, G.; Friedrich, A.W.; Markers, E.S. From theory to practice: Molecular strain typing for the clinical and public health setting. Euro. Surveill. 2013, 18, 20383. [Google Scholar] [CrossRef][Green Version]
- Kim, M.H.; Jeong, H.; Sim, Y.M.; Lee, S.; Yong, D.; Ryu, C.-M.; Choi, J.Y. Using comparative genomics to understand molecular features of carbapenem-resistant Acinetobacter baumannii from South Korea causing invasive infections and their clinical implications. PLoS ONE 2020, 15, e0229416. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, S.; Kim, M.H.; Kim, S.Y.; Nam, D.; Park, S.C.; Park, S.-H.; Bae, H.; Lee, H.-J.; Cho, J.H. Molecular epidemiology of carbapenem-resistant Acinetobacter baumannii isolates from a Korean hospital that carry blaOXA-23. Infect. Genet. Evol. 2018, 58, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Boolchandani, M.; D’Souza, A.W.; Dantas, G. Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 2019, 20, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Kumburu, H.H.; Sonda, T.L.; van Zwetselaar, M.; Leekitcharoenphon, P.; Lukjancenko, O.; Mmbaga, B.T.; Alifrangis, M.; Lund, O.; Aaresrup, F.; Kibiki, G. Using WGS to identify antibiotic resistance genes and predict antimicrobial resistance phenotypes in MDR Acinetobacter baumannii in Tazania. J. Antimicrob. Chemother. 2019, 74, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhu, Y.; Yi, Y.; Lu, N.; Zhu, B.; Hu, Y. Comparative genomic analysis of Acinetobacter baumannii clinical isolates reveals extensive genomic variation and diverse antibiotic resistance determinants. BMC Genom. 2014, 15, 1163. [Google Scholar] [CrossRef]
- Kwong, J.C.; McCallum, N.; Sintchenko, V.L.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef]
- Kumar, P.; Tech, B.; Sundermann, A.J.; Martin, E.; Snyder, G.M.; Marsh, J.W.; Harrison, L.H.; Roberts, M.S. Method for economic evaluation of bacterial whole genome sequencing surveillance compared to standard of care in detecting hospital outbreaks. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Dymond, A.; Davies, H.; Mealing, S.; Pollit, V.; Coll, F.; Brown, N.M.; Peacock, S.J. Genomic surveillance of methicillin-resistant Staphylococcus aureus: A mathematical early modelling study of cost effectiveness. Clin. Infect. Dis. 2020, 70, 1613–1619. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Specimen | Date of Isolation | Hospital Stay |
|---|---|---|---|
| #1 | TTA | 2015-01-20 | * |
| #2 | TTA | 2016-03-21 | ICU |
| #3 | Sputum | 2016-03-16 | ICU |
| #4 | Blood | 2016-04-13 | ICU |
| #5 | Keyboard | 2016-03-25 | ICU |
| #6 | Bed rail | 2016-03-28 | Ward A |
| #7 | Blood | 2015-01-20 | * |
| #8 | Blood | 2016-03-15 | ICU |
| #9 | Sputum | 2016-03-10 | ICU |
| #11 | Suction catheter | 2016-03-25 | ICU |
| #12 | Suction catheter | 2016-03-28 | Ward A |
| Isolate | ST | Beta-Lactam Resistance Genes | TZP | CAZ | FEP | IPM | MER | Aminoglycoside Resistance Genes | AMK | GEN | TOB | Sulfonamide Genes | SXT | Quinolone Genes | CIP | Phenicol Genes | Macrolide | Tetracycline Genes | CST |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | 191 | blaOXA-23, blaOXA-66, blaADC-25 | R | R | R | R | R | aadA1, aac(6′)-Ib-cr *, armA | R | R | R | sul1 | R | aac(6′)-Ib-cr | R | catB8 | msr(E). mph(E) | S | |
| #2 | 451 | blaOXA-23, blaOXA-66, blaADC-25, blaTEM-1D | R | R | R | R | R | aph(3′)-Ia, aph(6)-Id, aph(3″)-Ib, armA | R | R | R | sul2 | R | R | msr(E). mph(E) | tet(B) | S | ||
| #3 | 369 | blaOXA-23, blaOXA-66, blaADC-25 | R | R | R | R | R | aadA1, aac(6′)-Ib-cr *, armA | R | R | R | sul1 | R | aac(6′)-Ib-cr | R | catB8 | msr(E), mph(E) ** | S | |
| #4 | 369 | blaOXA-23, blaOXA-66, blaADC-25 | R | R | R | R | R | aadA1, aac(6′)-Ib-cr *, armA | R | R | R | sul1 | R | aac(6′)-Ib-cr | R | catB8 | msr(E). mph(E) | S | |
| #5 | 451 | blaOXA-23, blaOXA-66, blaADC-25, blaTEM-1D | R | R | R | R | R | aph(3′)-Ia, aph(6)-Id, aph(3″)-Ib, armA | R | R | R | sul2 | R | R | msr(E). mph(E) | tet(B) | S | ||
| #6 | 451 | blaOXA-23, blaOXA-66, blaADC-25, blaTEM-1D | R | R | R | R | R | aph(3′)-Ia, aph(6)-Id, aph(3″)-Ib, armA | R | R | R | sul2 | R | R | msr(E). mph(E) | tet(B) | S | ||
| #7 | 191 | blaOXA-23, blaOXA-66, blaADC-25 | R | R | R | R | R | aadA1, aac(6′)-Ib-cr *, armA | R | R | R | sul1 | S | aac(6′)-Ib-cr | R | catB8 | msr(E). mph(E) | S | |
| #8 | 369 | blaOXA-23, blaOXA-66, blaADC-25 | R | R | R | R | R | aadA1, aac(6′)-Ib-cr *, armA | R | R | R | sul1 | R | aac(6′)-Ib-cr | R | catB8 | msr(E), mph(E) ** | S | |
| #9 | 191 | blaOXA-23, blaOXA-66, blaADC-25 | R | R | R | R | R | aadA1, aac(6′)-Ib-cr *, armA, ant(3”)-Ia | R | R | R | sul1 | R | aac(6′)-Ib-cr | R | catB8 | msr(E). mph(E) | S | |
| #11 | 451 | blaOXA-23, blaOXA-66, blaADC-25, blaTEM-1D | R | R | R | R | R | aph(3′)-Ia, aph(6)-Id, aph(3″)-Ib, armA | R | R | R | sul2 | R | R | msr(E). mph(E) | tet(B) | S | ||
| #12 | 451 | blaOXA-23, blaOXA-66, blaADC-25, blaTEM-1D | R | R | R | R | R | aph(3′)-Ia, aph(6)-Id, aph(3″)-Ib, armA | R | R | R | sul2 | R | R | msr(E). mph(E) | tet(B) | S |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.M.; Cho, H.W.; Kim, T.Y.; Park, J.S.; Jung, J.; Song, K.-H.; Lee, H.; Kim, E.S.; Kim, H.B.; Park, K.U. Whole-Genome Sequencing for Investigating a Health Care-Associated Outbreak of Carbapenem-Resistant Acinetobacter baumannii. Diagnostics 2021, 11, 201. https://doi.org/10.3390/diagnostics11020201
Hwang SM, Cho HW, Kim TY, Park JS, Jung J, Song K-H, Lee H, Kim ES, Kim HB, Park KU. Whole-Genome Sequencing for Investigating a Health Care-Associated Outbreak of Carbapenem-Resistant Acinetobacter baumannii. Diagnostics. 2021; 11(2):201. https://doi.org/10.3390/diagnostics11020201
Chicago/Turabian StyleHwang, Sang Mee, Hee Won Cho, Tae Yeul Kim, Jeong Su Park, Jongtak Jung, Kyoung-Ho Song, Hyunju Lee, Eu Suk Kim, Hong Bin Kim, and Kyoung Un Park. 2021. "Whole-Genome Sequencing for Investigating a Health Care-Associated Outbreak of Carbapenem-Resistant Acinetobacter baumannii" Diagnostics 11, no. 2: 201. https://doi.org/10.3390/diagnostics11020201
APA StyleHwang, S. M., Cho, H. W., Kim, T. Y., Park, J. S., Jung, J., Song, K.-H., Lee, H., Kim, E. S., Kim, H. B., & Park, K. U. (2021). Whole-Genome Sequencing for Investigating a Health Care-Associated Outbreak of Carbapenem-Resistant Acinetobacter baumannii. Diagnostics, 11(2), 201. https://doi.org/10.3390/diagnostics11020201