1. Introduction
Emerging pathogens that cause severe infections and diseases present an urgent need for the development of novel, more rapid and sensitive methods and devices that allow for timely patient management. Despite continuous efforts encouraged by governments and health institutions worldwide for the development of point-of-care (PoC) diagnostic tools that allow the simultaneous detection and identification of pathogens, there is still no test that combines all advantages and attributes for a test of this type [
1,
2]. Traditional microbiology techniques used in the identification of infections and causative bacterial species always start by culturing the samples, followed by several molecular and biochemical tests, such as colony size and color, Gram stain, catalase/oxidase activity, coagulase test, etc. These techniques are precise and effective; however, they are time consuming, provide delayed information (2 to 3 days), and are expensive due to the need of trained personnel and specific lab equipment. Nowadays, PCR-based approaches and matrix-assisted laser desorption-ionization time of flight mass spectrometry (MALDI-TOF MS) methods are being used for their speed and sensitivity [
3,
4,
5,
6,
7,
8,
9]. However, amplification methods like PCR suffer from contamination problems, difficult experimental design, complex interpretation of results, and high costs. In the case of MALDI-TOF MS, the main limitation, besides the obvious high capital investment needed, is the inability to differentiate taxonomically related bacteria like high pathogenic species (e.g.,
Shigella) from commensal common bacteria (e.g.,
E. coli) and the need of an existing database that correlates the spectra with the pathogen [
7]. As a result, these techniques currently co-exist with bacterial cultures and a diagnostic result is obtained after 18–24 h or even longer [
8].
Consequently, there is an urgent and unmet clinical need for innovative PoC diagnostic tools that can reliably identify bacterial infections with short turnaround time, enabling doctors to treat patients with appropriate antibiotics as early as possible and to avoid the spread of bacteria and development of antibiotic resistance, especially in the hospital setting. The development of such rapid diagnostic methods will provide clinical and cost-related improvements in the management of infections.
Desirable features for the development of diagnostic devices should include simplicity of design, cost-effectiveness, portability, ease-of-use, no need for specialized personnel or complex laboratory instrumentation and short turn-around of results with high sensitivity and specificity. In the last decade, immunoassays have found wider acceptance due to their affordability, user friendliness and versatility of detection formats, making them an excellent choice for point-of-care applications [
10,
11]. Among all systems using antibodies as recognition molecules, lateral flow immuno-chromatography (LFIC) had recently achieved remarkably high level of impact, as it can be used for applications where fast and accurate detection is required [
12,
13], such as in the case of antibody or antigen tests for COVID-19. LFIC is a simple method of specifically detecting the presence or absence of a target analyte in a given sample matrix. LFIC is currently being used as a PoC tool (fast and in situ) in a large range of applications, from the widely known home pregnancy test to more specialized tests for clinical, food safety, environmental and industrial applications, eliminating the need for specialized and costly equipment and conventional laboratories [
14].
In our lab, we have taken advantage of the features of LFIC, and we sought to combine them with our extensive expertise in harnessing the biology of bacterial nucleases as biomarker of disease to create an approach that provides rapid and sensitive identification of bacterial infections. Bacteria are microorganisms that express a variety of enzymes and proteins which are unique for each species, even for members belonging to the same genus, thus making them useful tools for specific targeting. Nucleases are one type of these enzymes, responsible for many functions related to the regulation of bacteria genetic material by cleaving nucleic acids, such as DNA or RNA. Moreover, nucleic acids have been proven to be useful recognition molecules for the development of several diagnostic strategies, mostly due to their flexibility and adaptability to various transduction mechanisms, such as fluorescence, electrochemical, piezoelectric, and colorimetric [
15,
16]. In recent years, we have been working on various approaches based on nucleic acid substrates to target specific nucleases that could be used as a blueprint of bacterial infections or specific types of cancer [
17,
18,
19,
20]. The
Staphylococcus aureus (
S. aureus) bacteria, both the methicillin sensitive (MSSA) and methicillin resistant (MRSA) varieties, is the leading cause of nosocomial infections worldwide [
21,
22,
23,
24]. Therefore, fast, accurate identification of this bacteria is crucial to ensuring effective treatment and preventing the spread of infections within hospital settings.
In a previous report,
S. aureus infections were detected using an oligonucleotide fluorescent probe (TT probe) as a substrate that is specifically cleaved by the micrococcal nuclease (MN) produced by
S. aureus in less than 1 h [
20]. Herein, we propose to follow up on that study by translating this technology, based on nucleases and short nucleic acid (oligonucleotides) substrates, into an approach amenable to a portable and easy-to-use detection device based on a lateral flow system.
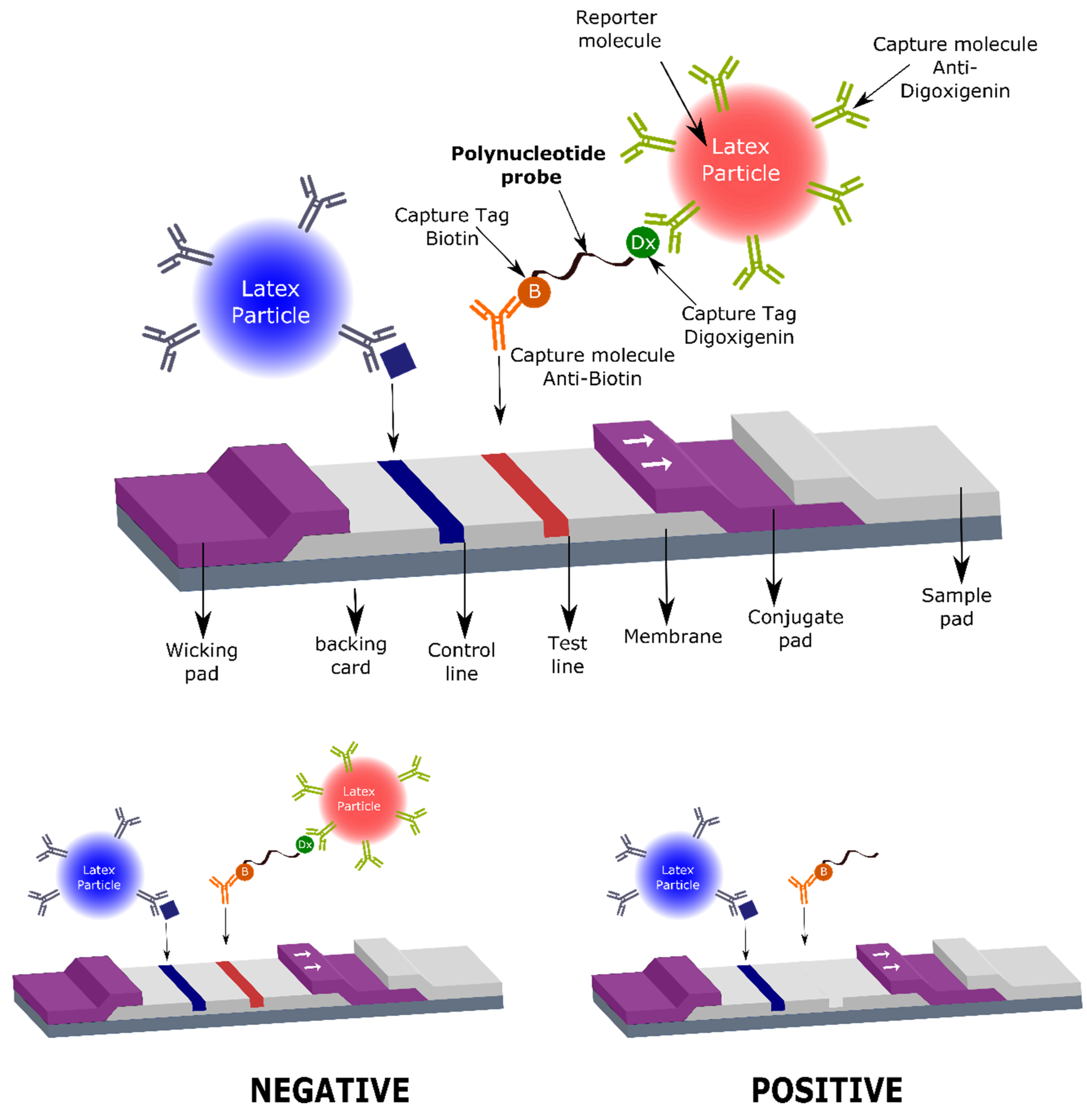
Therefore, herein we describe, for the first time, the utility of specific oligonucleotide probes, modified with tags that allow their capture on a lateral flow strip, for the rapid, sensitive, and specific detection of nuclease activity derived from
S. aureus, with a great potential for use in the clinical field. Specifically, the oligonucleotide probe that acts as substrate for the target nuclease is composed of a short oligonucleotide sequence flanked by two capture tags, digoxigenin and biotin (polynucleotide probe,
Figure 1), and we named it the LF-TTprobe. This oligonucleotide sequence consists of 2 central thymidines as the specific region, flanked by nucleosides modified with a methyl group at the 2′-position of the sugar ribose (2′-O methyl), that renders the probe resistant to non-specific nucleases.
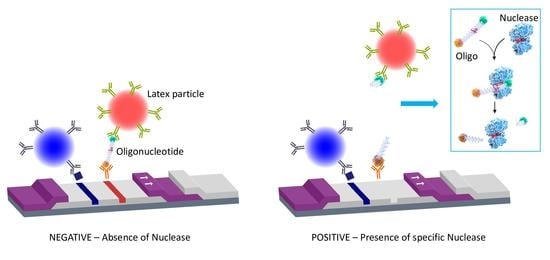
The digoxigenin tag (Dx) binds a reporter molecule (latex microparticle, red), previously functionalized with anti-digoxigenin antibodies, which resides in the conjugate pad of the strip. On the other hand, the anti-biotin molecule captures the nucleic acid probe in the test line of the strip by the other end, where the biotin (B) is attached. If the target nuclease (e.g.,
S. aureus nuclease) is absent, the detection complex formed by the nucleic acid probe and the red latex microparticle is visualized as a red line by accumulation of the red latex particle complexes in the test line area (negative result,
Figure 1 lower left panel). If the target nuclease (e.g.,
S. aureus nuclease) is present, the nucleic acid probe is cleaved, releasing the oligonucleotide fragment along with the digoxigenin tag which is complexed to the red latex microparticle. In this situation, no test line is detected, thus indicating the presence of nucleases in the biological sample. Consequently, a positive result in this system shows no test line (
Figure 1, lower right panel).
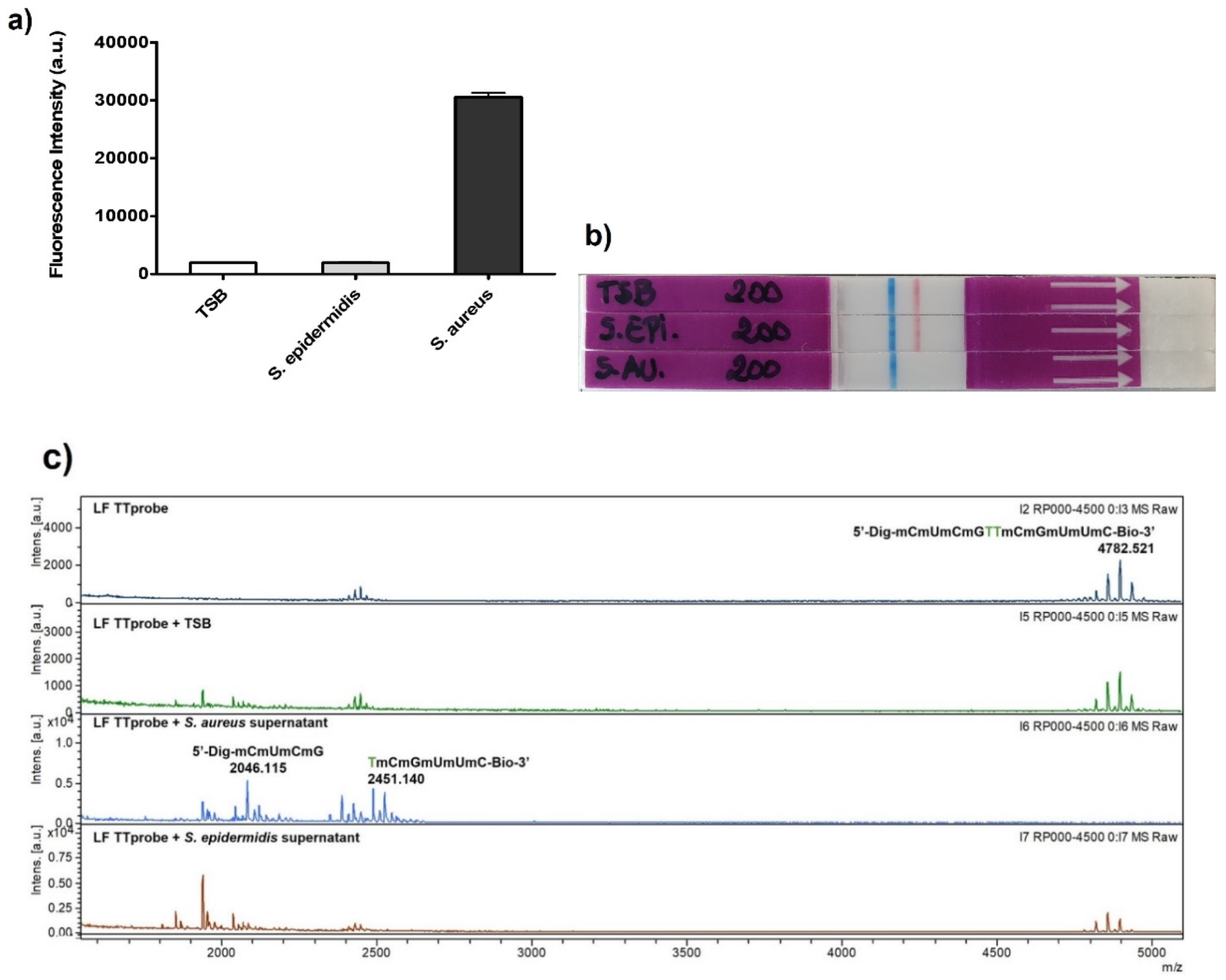
The LFIC system performance was tested using culture supernatants of
S. aureus as the target pathogen, and culture supernatants of
Staphylococcus epidermidis (
S. epidermidis) as the control pathogen. The choice of the control bacterium was dictated by two factors. On one hand,
S. epidermidis is the main bacterium present in clinical samples that might show cross-reactivity with
S. aureus. On the other hand, both bacteria are endowed with similar surface-expressed proteins and secrete virulence factors such as toxins, and enzymes such as nucleases. However, when compared to
S. aureus,
S. epidermidis presents a smaller repertoire of nucleases [
25], thus enabling technologies based on nucleases to discriminate between them. Our technology, presented herein, is based on harnessing the activity of a specific secreted nuclease, allowing for their differentiation. Moreover, both
S. aureus and
S. epidermidis are also clearly differentiated from other common bacteria causing severe infections, such as
Klebsiella pneumoniae,
Pseudomonas aeruginosa,
Escherichia coli and
Streptococcus pneumoniae, showing the high specificity of the method.
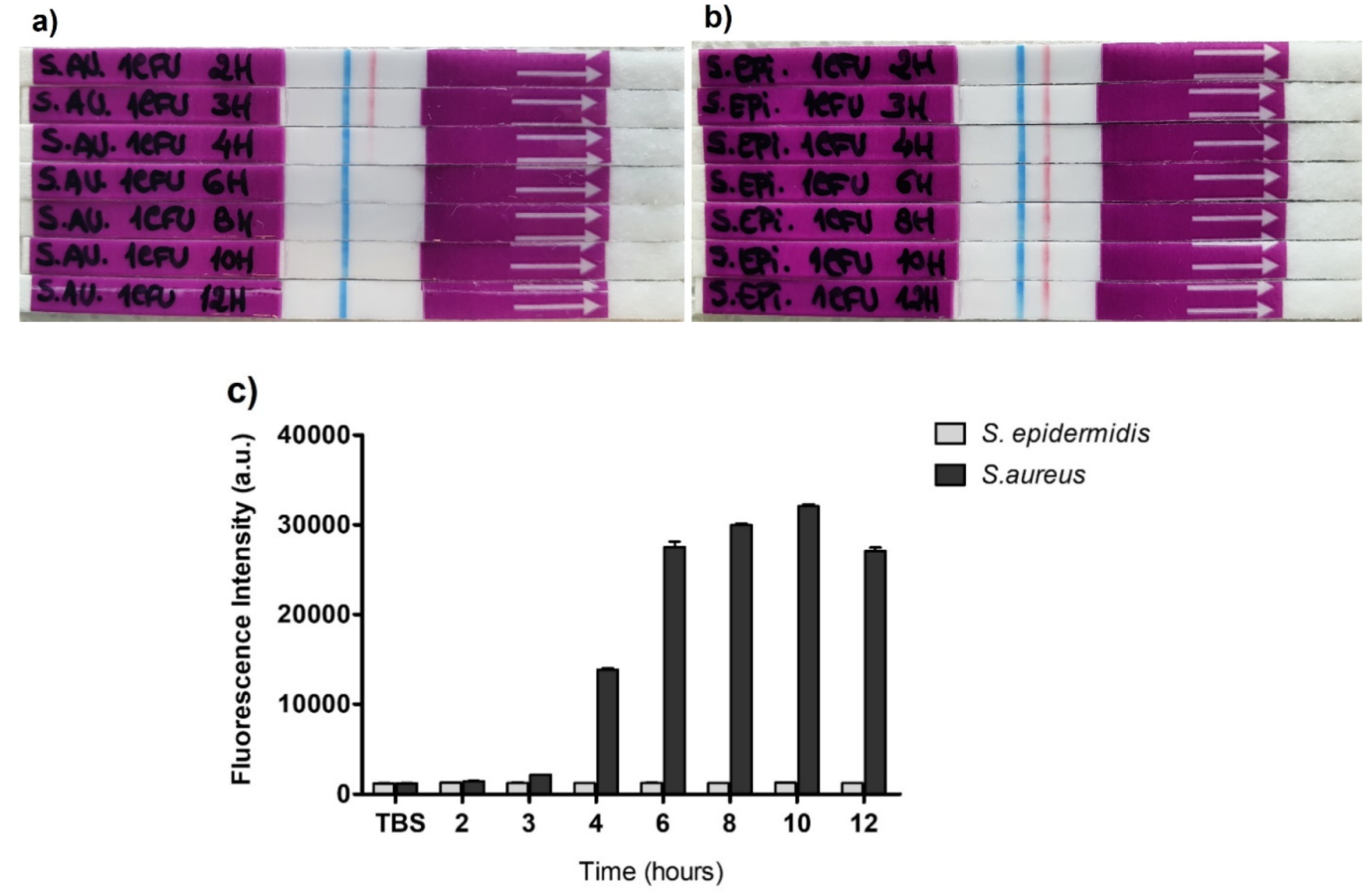
In this contribution, we demonstrate a significant improvement in the sensitivity of S. aureus detection when compared to the previously described fluorescent approach by achieving a 250-fold reduction in the amount of the oligonucleotide probe used for the optimal detection of its target nuclease, hence decreasing the potential cost of LFIC device manufacturing. Importantly, we show that we can detect the presence of S. aureus via its secreted nuclease activity in mini cultures of 6 h, obtained from one colony forming unit (CFU) culture, thus significantly shortening the actual waiting time of the gold standard method, which is usually between 18 to 24 h. This unprecedented three-in-one feature, increased sensitivity (S), specificity (S) and speed (S), has paramount implications on the development of LF applications for clinical diagnostics, with great potential to be extended to other pathogens and human conditions.
2. Materials and Methods
2.1. Buffer Solutions and Culture Media
Tris-EDTA, 1X solution (TE, CAS number: 38641-82-6, pH 8.0) was purchased from Thermo Fisher Scientific (Madrid, Spain). Tris buffered saline (TBS) purchased from Merck Life Science (Madrid, Spain) was prepared by dissolving the powder of a pHast pack in 500 mL of ultrapure water to obtain a solution of 1× with final concentrations of 50 mM Tris, 138 mM NaCl, 2.7 mM KCl, pH 8.0. Tween 20 was purchased from Sigma-Aldrich and used as received to prepare the running buffers for the lateral flow strips at a final concentration of 0.05% V/V. Calcium chloride (CAS number: 1043-52-4) purchased from Merck Life Science (Madrid, Spain) was prepared in ultrapure water at a final concentration of 100 mM. Phosphate buffered saline 10× (PBS, pH 7.4) without MgCl2 and CaCl2 (PBS-/-) was purchased from Thermo Fisher Scientific (Madrid, Spain). This solution was used at 1× by diluting it ten times in ultrapure water. Ethylenediamine tetraacetate acid (EDTA) 500 mL, 0.5 M, was purchased from Thermo Fisher Scientific (Madrid, Spain). PBS-/- with EDTA solution was prepared using PBS-/- 1× and EDTA 500 mM for a final concentration of 10 mM EDTA.
Tryptic soy broth (TSB), nutrient broth (NB), Todd Hewitt broth (THB) culture media, yeast extract and European bacteriological agar were purchased from Laboratorios Conda SL (Madrid, Spain) and prepared in ultrapure water, as indicated. The agar was always used by mixing 15 g per liter prepared with the broth for incubation in Petri dishes. All the media were always autoclaved at 121 °C for 25 min.
2.2. Bacterial Strains and 24 h Supernatant Preparation
All the bacterial strains used in this study were obtained from LGC Standards (Barcelona, Spain) and cultured using the culture conditions recommended by the American Type Culture Collection (ATCC).
Table 1 details all bacteria strains used herein, with their respective culture media.
To prepare pure culture supernatants, frozen bacteria stored at −80 °C were pre-cultured in each correspondent medium (
Table 1) for 18 h at 37 °C with agitation at 180 rpm. In the case of
Streptococcus pneumoniae, the culture was always supplemented with 5% CO
2. One milliliter of each bacterium in glycerol solution was added to 50 mL of the correspondent broth medium. After 18 h of growth, a dilution of 1:500 was prepared by adding 100 μL of the 18 h pre-culture to 50 mL of fresh medium (sub-culture). Using 10 μL disposable loops, Petri dishes with the corresponding agar were also prepared to obtain isolated colonies. Both, broth and dish preparations were incubated for 24 h at 37 °C with and without agitation at 180 rpm, respectively.
After 24 h of growth, each culture was then centrifuged (6000× g for 20 min) and the supernatant was removed and stored at −20 °C until use. The pellets were properly discarded in biohazard waste. The Petri dishes were stored at 4 °C until use.
2.3. Oligonucleotide Probes Synthesis and Purification
The probe was synthesized and purified at Biomers.net (Ulm, Germany). The oligonucleotide sequence of TTprobe was the following: 5′-mCmUmCmGdTdTmCmGmUmUmC-3′ (m = -methyl at 2′; d = DNA nucleoside with no modification). The TTprobe for the lateral flow tests contained a digoxigenin molecule at the 5′-end and a biotin molecule at the 3′-end (LF-TTprobe). For this, a standard method of solid phase phosphoramidite chemistry was used, followed by high-performance liquid chromatography (HPLC) purification. The probe identity was confirmed by MALDI-MS. The purity of the probe, as assessed by HPLC analysis, is typically greater than 95%.
The TTprobe used in the fluorescence experiments (F-TTprobe) was synthetized with a fluorophore and a quencher, as previously reported [
20].
2.4. Oligonucleotide Probes Solution Preparation
The oligonucleotide probe stock solutions were prepared by resuspending the lyophilized probe vial (from the manufacturer) in TE buffer at a final concentration of 500 pmol·μL−1. Then, as needed, working solutions of the fluorescent probe were prepared by dissolving the F-TTprobe in PBS-/-. To prepare working solutions of the lateral flow probe, the LF-TTprobe was resuspended in ultrapure water. Note that probe solutions prepared in PBS are not recommended for lateral flow assays.
2.5. Lateral Flow Strips
The lateral flow strips used throughout this work were purchased from Operon (Cuarte de Huerva, Zaragoza, Spain) and Abingdon Health (Sand Hutton, York, UK). The Operon strips use latex particles as reporter molecules, while Abingdon PCR-Flex strips use carbon particles as reporters. Both strips contain anti-digoxigenin/biotin antibodies.
2.5.1. Optimization of the Strips—Running Buffer
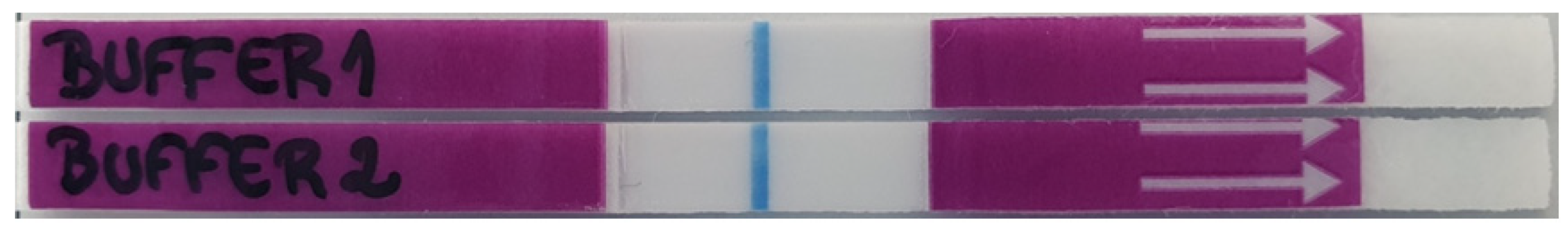
To better visualize the strip lines for an accurate reading of the lateral flow (LFA) results, an appropriate running buffer should be used. The running buffer should not only maintain the sample pH, but also allow a perfect flow through the strip, and not interfere with the antigen–antibody interactions. Therefore, we tested and evaluated the performance of two buffers: TBS buffer alone (Buffer 1) and TBS buffer supplemented with tween 20 0.05% v/v (Buffer 2).
2.5.2. Optimization of the Strips—Probe Concentration
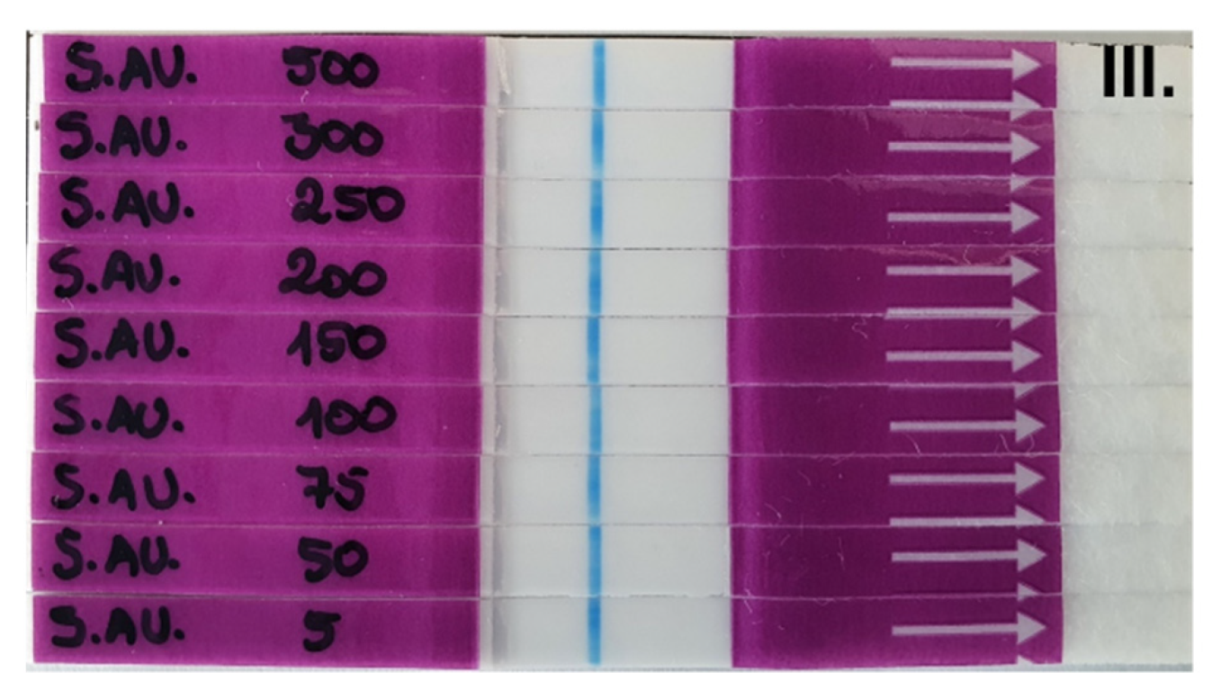
To determine the best concentration of probe to be used in the LF system, a titration with different concentrations of the LF-TTprobe was performed, using S. aureus ATCC 29213 supernatants as the sample and TSB medium and the S. epidermidis ATCC 35984 supernatant as the control. LF-TTprobe solutions at different concentrations (500, 300, 250, 200, 150, 100, 75, 50 and 5 fmol·μL−1) were prepared in ultrapure water.
2.6. Nuclease Activity Assays (NAA)
2.6.1. Fluorescence Detection
The fluorescence nuclease activity assays were performed in reaction volumes of 10 μL, by mixing 8 μL of sample (24 h culture supernatants or control medium) with 1 μL of CaCl2 100 mM and 1 μL of F-TTprobe (50 pmol·μL−1). The reactions were incubated at 37 °C, for 30 min. Then, the reactions were stopped by adding 295 mL of PBS-/- supplemented with 10 mM EDTA. Finally, 95 mL of each sample was loaded in triplicate into 96-well black plates (96F non-treated black microwell plate, Thermo Scientific). Fluorescence intensity was measured with a fluorescence microplate reader, Synergy Neo2 Hybrid Multi-Mode Reader from Agilent BioTek (Winooski, VT, USA) using the filter settings for FAM (excitation/emission 485/528 nm). Three independent experiments were performed with each sample. The results are expressed as the mean ± SD (n = 3) of fluorescence intensity in arbitrary units (a.u.).
2.6.2. Lateral Flow Detection
The nuclease activity assays for the lateral flow detection were prepared in a similar fashion as for the fluorescence assays, by only changing the probe and probe concentration. Therefore, 8 μL of each sample (24 h culture supernatants or control medium), were mixed with 1 μL of CaCl2 100 mM and 1 μL of LF-TTprobe (200 fmol·μL−1), to a final reaction volume of 10 μL. Then, the reactions were incubated at 37 °C for 30 min. Next, to allow the reaction to flow through the strip, 150 μL of TBS + tween 20 0.05% running buffer were added to the reaction. Thus, a final volume of 160 μL was transferred to a 96-well plate with a flat bottom so toallow the strips, when dipped in the wells, to be in contact with the entire volume of the sample. Then the strips were labeled, dipped in the correspondent well and the flow was allowed to run for 10 min. Next, the strips were removed from the wells and the results were visually read. Photographs of the strips were also taken and recorded.
2.6.3. MALDI-TOF
For an optimal MALDI evaluation, the NAA was slightly modified from the original version. Thus, 1 μL of sample, S. aureus ATCC 29213, S. epidermidis ATCC 35984 or TSB as control, was mixed with 1 μL of CaCl2 100 mM and 1 μL of LF-TTprobe (with digoxigenin and biotin) and prepared in ultrapure water at a concentration of 50 pmol·μL−1 (final concentration 5 pmol·μL−1) in 7 μL of ultrapure water and incubated at 37 °C for 30 min. Next, the reaction was not stopped, but was directly used for crystallization. The sample preparation was carried out by the deposition of 0.5 μL of the sample (NAA reaction) directly onto a polished stainless-steel plate (Bruker Daltonics, Bremen, Germany), and mixing it with 0.5 μL of matrix solution, which was composed of a dissolution of 10 mg of 2,4,6-trihydroxyacetophenone (THAP) and 5 mg of diammonium citrate in 1 mL of deionized water. MALDI-TOF MS analysis was performed using an UltrafleXtreme III time-of-flight mass spectrometer equipped with a Nd:YAG laser (Smartbeam II, 355 nm, 1 kHz), controlled by Flex Control 3.3 software (Bruker Daltonics, Bremen, Germany). The acquisitions were carried out in positive-ion linear mode at a laser frequency of 1 kHz. The spectrum was acquired at 30% laser fluency and was recorded in the m/z range from 1500 to 5000. The deflector cut-off was set at m/z 1300, and the spectrum resulted from the accumulation of 3000 laser shots. All the spectra were analyzed using FlexAnalysis software 3.0 (Bruker Daltonics, Bremen, Germany).
2.7. Sensitivity Tests
The sensitivity experiments were carried out using supernatants of
S. aureus ATCC 29213 and
S. epidermidis ATCC 35984. Tenfold serial dilutions, from 10
−1 down to 10
−6, of these supernatants were prepared in ultrapure water. Next, NAA for lateral flow was performed as previously described in
Section 2.6.2.
2.8. Specificity Tests
The specificity experiments were carried out using supernatants of all bacteria detailed in
Table 1,
Section 2.2. The NAA for lateral flow was performed as previously described in
Section 2.6.2.
2.9. Short Cultures of S. aureus ATCC 29213 and S. epidermidis ATCC 35984
Cultures of
S. aureus ATCC 29213 and
S. epidermidis ATCC 35984 were prepared from 1 CFU taken from previously prepared Petri dishes that were stored at 4 °C, as detailed in
Section 2.2.
One CFU of each bacterium was added to 50 mL of TSB culture medium using a collection loop and incubated at 37 °C with 180 rpm agitation. Next, various samples were collected at the following incubation times: 2 h, 3 h, 4 h, 6 h, 8 h, 10 h and 12 h. One milliliter of each bacterial culture was collected and centrifuged at 13400 rpm for 20 min. The supernatants were then kept at 4 °C until use in the NAA. The pellets were properly discarded. Before proceeding with the NAA, all the samples to be used were prepared by mixing 10 μL of each sample with 10 μL of TBS buffer (1:1) (pre-NAA prep.). The NAAs were performed for fluorescence and lateral flow detection as explained in
Section 2.6.1 and
Section 2.6.2, using as a sample 8 μL of each pre-NAA prep.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}