
Mutational Analysis and mtDNA Haplogroup Characterization in Three Serbian Cases of Mitochondrial Encephalomyopathies and Literature Review

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Ethical Considerations
2.3. Molecular Genetic Methods
2.4. Haplogroups Analysis and Phylogenetic Tree Reconstruction
2.5. Bioinformatics Analysis
3. Results
3.1. Mutational Genetic Analysis
3.2. MITOMASTER Analysis
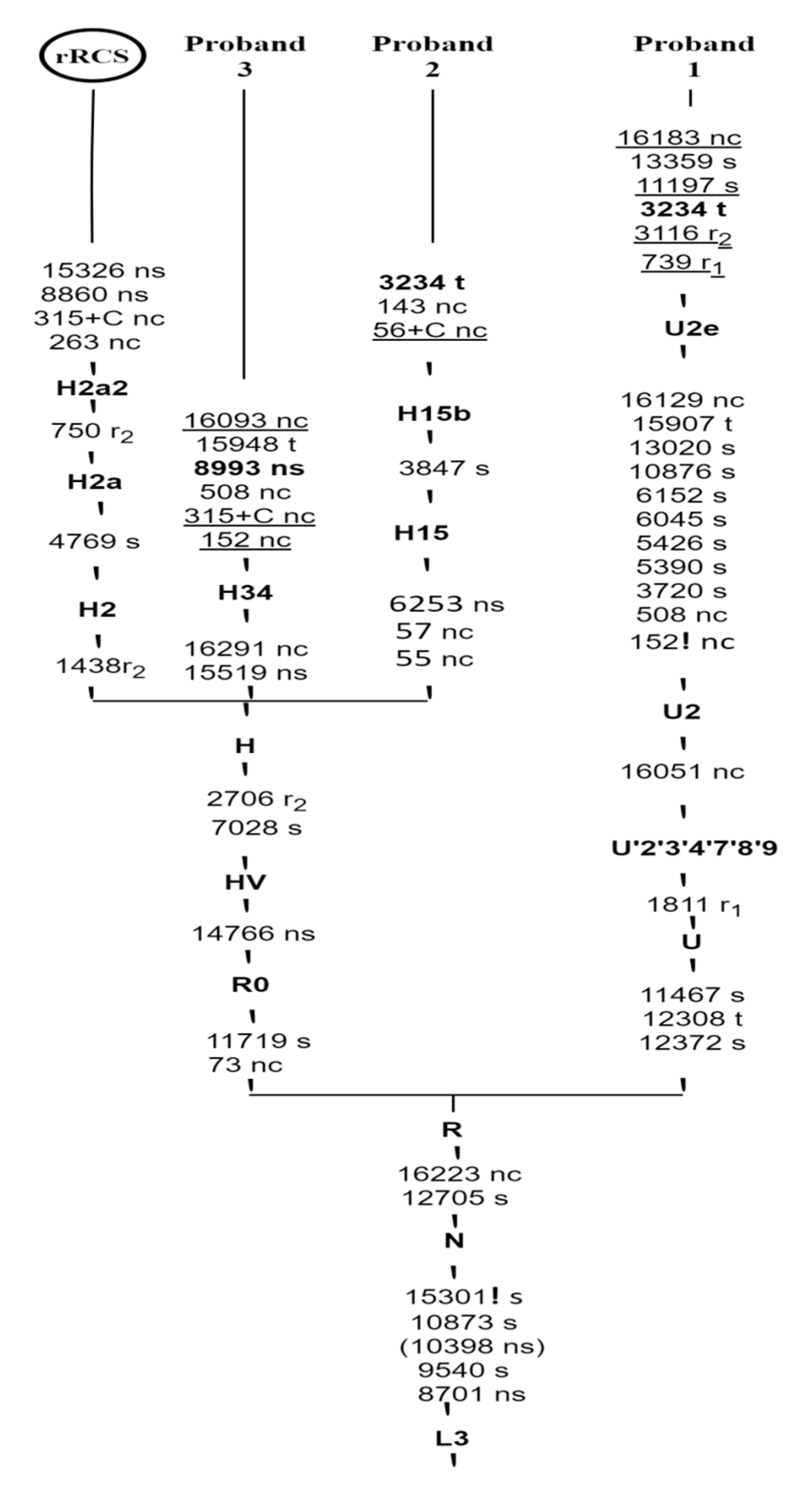
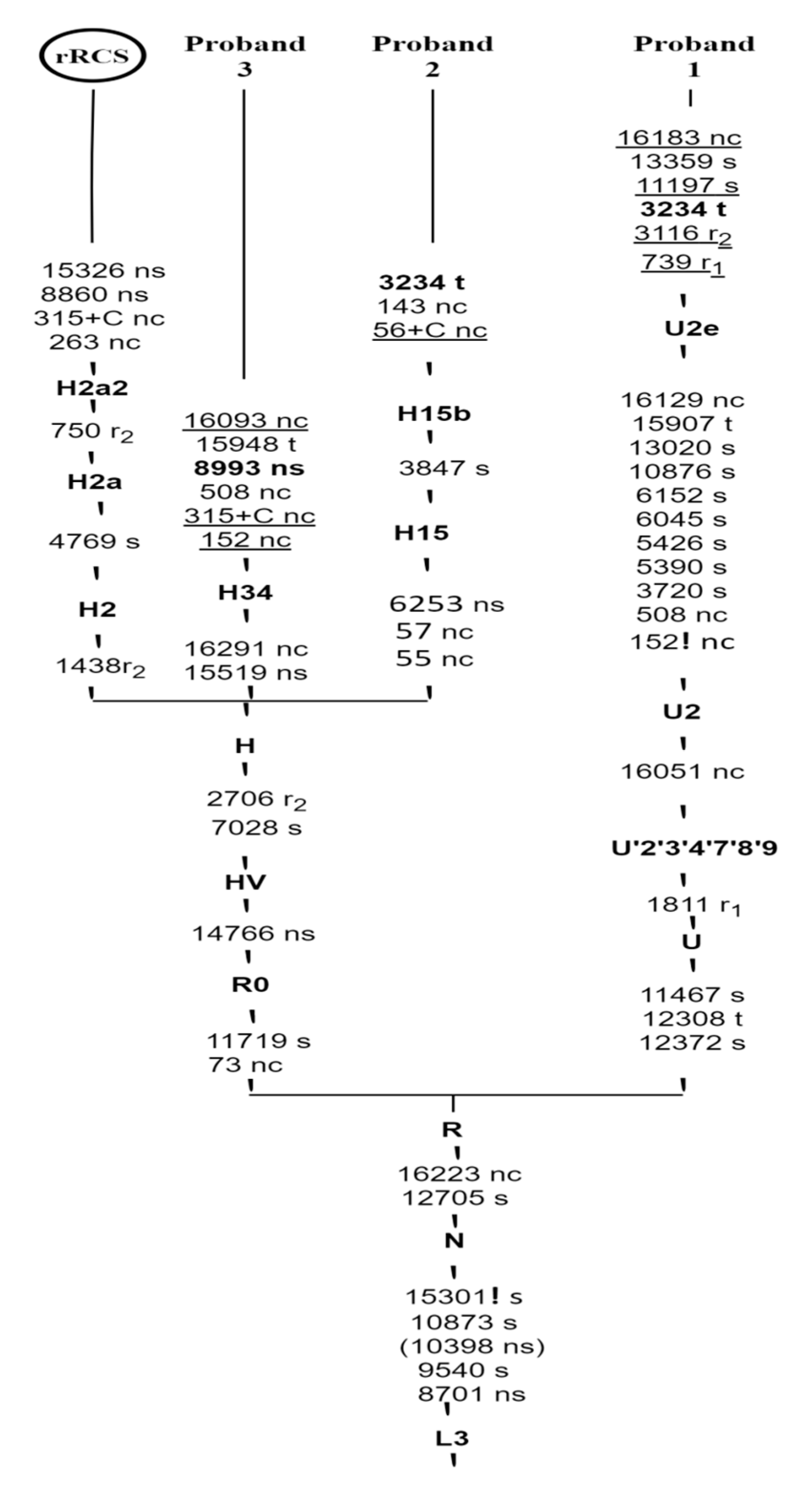
3.3. Phylogenetic Tree Construction
3.4. Genotype-Phenotype Relationship
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nyhan, W.L.; Hoffmann, G.F.; Hoffmann, G.F. Atlas of Inherited Metabolic Diseases, 4th ed.; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Sperl, W. [Diagnosis and therapy of mitochondriopathies]. Wien. Klin. Wochenschr. 1997, 109, 93–99. [Google Scholar] [PubMed]
- Bugiardini, E.; Mitchell, A.L.; Rosa, I.D.; Horning-Do, H.-T.; Pitmann, A.; Poole, O.V.; Holton, J.L.; Shah, S.; Woodward, C.; Hargreaves, I.; et al. MRPS25 Mutations Impair Mitochondrial Translation and Cause Encephalomyopathy. Hum. Mol. Genet. 2019, 28, 2711–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colina-Tenorio, L.; Horten, P.; Pfanner, N.; Rampelt, H. Shaping the Mitochondrial Inner Membrane in Health and Disease. J. Intern. Med. 2020, 287, 645–664. [Google Scholar] [CrossRef]
- Rahman, S.; Wolf, N.I. Diagnostic Workup of Patients with Mitochondrial Diseases. In Inherited Metabolic Diseases; Hoffmann, G., Zschocke, J., Nyhan, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 521–535. [Google Scholar]
- DiMauro, S.; Moraes, C.T. Mitochondrial Encephalomyopathies. Arch. Neurol. 1993, 50, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Valenzuela, P.L.; Laine-Menéndez, S.; Fernández-de la Torre, M.; Bermejo-Gómez, V.; Rufián-Vázquez, L.; Arenas, J.; Martín, M.A.; Lucia, A.; Morán, M. Physical Exercise and Mitochondrial Disease: Insights from a Mouse Model. Front. Neurol. 2019, 10, 790. [Google Scholar] [CrossRef] [PubMed]
- Roberta, C.; Falchetti, A. Mitochondriopathies and Bone Health. Tre. Bio. Res. 2018, 1, 1–7. [Google Scholar] [CrossRef]
- Swerdlow, R.H. The Neurodegenerative Mitochondriopathies. J. Alzheimers Dis. 2009, 17, 737–751. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, T.G.; Suomalainen, A. Mitochondrial DNA Can Be Inherited from Fathers, Not Just Mothers. Nature 2019, 565, 296. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F. Primary Mitochondrial Disorders Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- de Laat, P.; Janssen, M.C.H.; Alston, C.L.; Taylor, R.W.; Rodenburg, R.J.T.; Smeitink, J.A.M. Three Families with ‘de Novo’ m.3243A > G Mutation. BBA Clin. 2016, 6, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Poulton, J.; Finsterer, J.; Yu-Wai-Man, P. Genetic Counselling for Maternally Inherited Mitochondrial Disorders. Mol. Diagn. Ther. 2017, 21, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Juettner, R.; Zink, A.; Mlody, B.; Glazar, P.; Secker, C.; Ciptasari, U.H.; Stenzel, W.; et al. SURF1 Mutations Causative of Leigh Syndrome Impair Human Neurogenesis. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Shiraishi, H.; Egawa, K.; Uchida, M.; Ueno, M. Efficacy of Perampanel for Epileptic Seizures and Daily Behavior in a Patient with Leigh Syndrome: A Case Report. Brain Dev. 2021, 43, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh Syndrome: One Disorder, More than 75 Monogenic Causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Sofou, K.; Kollberg, G.; Hedberg-Oldfors, C.; Oldfors, A. The Phenotypic Variability and Natural History of NARS2 Associated Disease. Eur. J. Paediatr. Neurol. 2021, 31, 31–37. [Google Scholar] [CrossRef]
- Torraco, A.; Nasca, A.; Verrigni, D.; Pennisi, A.; Zaki, M.S.; Olivieri, G.; Assouline, Z.; Martinelli, D.; Maroofian, R.; Rizza, T.; et al. Novel NDUFA12 Variants Are Associated with Isolated Complex I Defect and Variable Clinical Manifestation. Hum. Mutat. 2021, 42, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A New Mitochondrial Disease Associated with Mitochondrial DNA Heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Saneto, R.P.; Patrick, K.E.; Perez, F.A. Homoplasmy of the m. 8993 T>G Variant in a Patient without MRI Findings of Leigh Syndrome, Ataxia or Retinal Abnormalities. Mitochondrion 2021, 59, 58–62. [Google Scholar] [CrossRef]
- de Vries, D.D.; van Engelen, B.G.; Gabreëls, F.J.; Ruitenbeek, W.; van Oost, B.A. A Second Missense Mutation in the Mitochondrial ATPase 6 Gene in Leigh’s Syndrome. Ann. Neurol. 1993, 34, 410–412. [Google Scholar] [CrossRef]
- Sgarbi, G.; Baracca, A.; Lenaz, G.; Valentino, L.M.; Carelli, V.; Solaini, G. Inefficient Coupling between Proton Transport and ATP Synthesis May Be the Pathogenic Mechanism for NARP and Leigh Syndrome Resulting from the T8993G Mutation in MtDNA. Biochem. J. 2006, 395, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Ruhoy, I.S.; Saneto, R.P. The Genetics of Leigh Syndrome and Its Implications for Clinical Practice and Risk Management. Appl. Clin. Genet. 2014, 7, 221–234. [Google Scholar]
- Uziel, G.; Ghezzi, D.; Zeviani, M. Infantile Mitochondrial Encephalopathy. Semin. Fetal Neonatal Med. 2011, 16, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Bonilla, E.; DiMauro, S. Mitochondrial DNA Mutations and Pathogenesis. J. Bioenerg. Biomembr. 1997, 29, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Suzuki, T.; Ishii, N.; Ohta, S.; Watanabe, K. Wobble Modification Defect in TRNA Disturbs Codon–Anticodon Interaction in a Mitochondrial Disease. EMBO J. 2001, 20, 4794–4802. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Horai, S.; Matsuoka, T.; Koga, Y.; Nihei, K.; Kobayashi, M.; Nonaka, I. Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like Episodes (MELAS): A Correlative Study of the Clinical Features and Mitochondrial DNA Mutation. Neurology 1992, 42, 545. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Pearce, S.F.; Powell, C.A.; D’Souza, A.R.; Nicholls, T.J.; Minczuk, M. Mitochondrial Transcript Maturation and Its Disorders. J. Inherit. Metab. Dis. 2015, 38, 655–680. [Google Scholar] [CrossRef] [Green Version]
- Queen, R.A.; Steyn, J.S.; Lord, P.; Elson, J.L. Mitochondrial DNA Sequence Context in the Penetrance of Mitochondrial T-RNA Mutations: A Study across Multiple Lineages with Diagnostic Implications. PLoS ONE 2017, 12, e0187862. [Google Scholar] [CrossRef] [Green Version]
- Al Yazidi, G.; Mulder, J.; Licht, C.; Harvey, E.; Robertson, J.; Sondheimer, N.; Tein, I. Reversal of Stroke-Like Episodes With L-Arginine and Meticulous Perioperative Management of Renal Transplantation in a Patient with Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-Like Episodes (MELAS) Syndrome. Case Report. Neurohospitalist 2021. [Google Scholar] [CrossRef]
- Montagna, P.; Gallassi, R.; Medori, R.; Govoni, E.; Zeviani, M.; Mauro, S.D.; Lugaresi, E.; Andermann, F. MELAS Syndrome: Characteristic Migrainous and Epileptic Features and Maternal Transmission. Neurology 1988, 38, 751. [Google Scholar] [CrossRef]
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes: A Distinctive Clinical Syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef]
- Caporali, L.; Maresca, A.; Capristo, M.; Del Dotto, V.; Tagliavini, F.; Valentino, M.L.; La Morgia, C.; Carelli, V. Incomplete Penetrance in Mitochondrial Optic Neuropathies. Mitochondrion 2017, 36, 130–137. [Google Scholar] [CrossRef]
- Carelli, V.; Achilli, A.; Valentino, M.L.; Rengo, C.; Semino, O.; Pala, M.; Olivieri, A.; Mattiazzi, M.; Pallotti, F.; Carrara, F.; et al. Haplogroup Effects and Recombination of Mitochondrial DNA: Novel Clues from the Analysis of Leber Hereditary Optic Neuropathy Pedigrees. Am. J. Hum. Genet. 2006, 78, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Georgiou, A.; Demetriou, C.A.; Heraclides, A.; Christou, Y.P.; Leonidou, E.; Loukaides, P.; Yiasoumi, E.; Panagiotou, D.; Manoli, P.; Thomson, P.; et al. Mitochondrial Superclusters Influence Age of Onset of Parkinson’s Disease in a Gender Specific Manner in the Cypriot Population: A Case-Control Study. PLoS ONE 2017, 12, e0183444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridge, P.G.; Maxwell, T.J.; Corcoran, C.D.; Norton, M.C.; Tschanz, J.T.; O’Brien, E.; Kerber, R.A.; Cawthon, R.M.; Munger, R.G.; Kauwe, J.S.K. Mitochondrial Genomic Analysis of Late Onset Alzheimer’s Disease Reveals Protective Haplogroups H6A1A/H6A1B: The Cache County Study on Memory in Aging. PLoS ONE 2012, 7, e45134. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chinnery, P.F. Inheritance of Mitochondrial DNA in Humans: Implications for Rare and Common Diseases. J. Intern. Med. 2020, 287, 634–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, G.P.H. Iatrogenic Mitochondriopathies: A Recent Lesson from Nucleoside/Nucleotide Reverse Transcriptase Inhibitors. In: Scatena R., Bottoni P., Giardina B. (eds) Advances in Mitochondrial Medicine. Adv. Exp. Med. Biol. 2012, 942, 347–369. [Google Scholar]
- Lorenzo, C.D.; Pierelli, F.; Coppola, G.; Grieco, G.S.; Rengo, C.; Ciccolella, M.; Magis, D.; Bolla, M.; Casali, C.; Santorelli, F.M.; et al. Mitochondrial DNA Haplogroups Influence the Therapeutic Response to Riboflavin in Migraineurs. Neurology 2009, 72, 1588–1594. [Google Scholar] [CrossRef]
- Torroni, A.; Campos, Y.; Rengo, C.; Sellitto, D.; Achilli, A.; Magri, C.; Semino, O.; García, A.; Jara, P.; Arenas, J.; et al. Mitochondrial DNA Haplogroups Do Not Play a Role in the Variable Phenotypic Presentation of the A3243G Mutation. Am. J. Hum. Genet. 2003, 72, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Kong, Q.; Zhang, Y. Application of the Phylogenetic Analysis in Mitochondrial Disease Study. Sci. Bull. 2008, 53, 2733–2738. [Google Scholar] [CrossRef] [Green Version]
- Makino, M.; Horai, S.; Goto, Y.; Nonaka, I. Mitochondrial DNA Mutations in Leigh Syndrome and Their Phylogenetic Implications. J. Hum. Genet. 2000, 45, 69–75. [Google Scholar] [CrossRef]
- Taylor, R.W.; Taylor, G.A.; Durham, S.E.; Turnbull, D.M. The Determination of Complete Human Mitochondrial DNA Sequences in Single Cells: Implications for the Study of Somatic Mitochondrial DNA Point Mutations. Nucleic Acids Res. 2001, 29, e74. [Google Scholar] [CrossRef]
- Dawod, P.G.A.; Jancic, J.; Marjanovic, A.; Brankovic, M.; Jankovic, M.; Samardzic, J.; Potkonjak, D.; Djuric, V.; Mesaros, S.; Novakovic, I.; et al. Whole Mitochondrial Genome Analysis in Serbian Cases of Leber’s Hereditary Optic Neuropathy. Genes 2020, 11, 1037. [Google Scholar] [CrossRef]
- Brandon, M.C.; Ruiz-Pesini, E.; Mishmar, D.; Procaccio, V.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Patil, U.; Baldi, P.; Wallace, D.C. MITOMASTER—A Bioinformatics Tool for the Analysis of Mitochondrial DNA Sequences. Hum. Mutat. 2009, 30, 1–6. [Google Scholar] [CrossRef] [Green Version]
- van Oven, M. PhyloTree Build 17: Growing the Human Mitochondrial DNA Tree. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e392–e394. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and Revision of the Cambridge Reference Sequence for Human Mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An Enhanced MITOMAP with a Global MtDNA Mutational Phylogeny. Nucleic Acids Res. 2007, 35, D823–D828. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER Version 11: Expanded Annotation Data from Gene Ontology and Reactome Pathways, and Data Analysis Tool Enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the Pathogenicity of Novel Variants in Mitochondrial TRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef] [PubMed]
- Pütz, J.; Dupuis, B.; Sissler, M.; Florentz, C. Mamit-TRNA, a Database of Mammalian Mitochondrial TRNA Primary and Secondary Structures. RNA 2007, 13, 1184–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of Nuclear and Mitochondrial DNA Mutations Related to Adult Mitochondrial Disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Y.S.; DiMauro, S.; Turnbull, D.M. Mitochondrial Medicine: A Historical Point of View. In Diagnosis and Management of Mitochondrial Disorders; Mancuso, M., Klopstock, T., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–18. [Google Scholar]
- Sharma, R.; Reinstadler, B.; Engelstad, K.; Skinner, O.S.; Stackowitz, E.; Haller, R.G.; Clish, C.B.; Pierce, K.; Walker, M.A.; Fryer, R.; et al. Circulating Markers of NADH-Reductive Stress Correlate with Mitochondrial Disease Severity. J. Clin. Investig. 2021, 131, e136055. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Thorburn, D. Nuclear Gene-Encoded Leigh Syndrome Spectrum Overview. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2015; [Updated 2020 July 16]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK320989/ (accessed on 31 August 2021).
- Thorburn, D.R.; Rahman, J.; Rahman, S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zhang, H.; Burr, S.P.; Chinnery, P.F. The Mitochondrial DNA Genetic Bottleneck: Inheritance and Beyond. Essays Biochem. 2018, 62, 225–234. [Google Scholar]
- Fujii, T.; Hattori, H.; Higuchi, Y.; Tsuji, M.; Mitsuyoshi, I. Phenotypic Differences between T-->C and T-->G Mutations at Nt 8993 of Mitochondrial DNA in Leigh Syndrome. Pediatr. Neurol. 1998, 18, 275–277. [Google Scholar] [CrossRef]
- Weerasinghe, C.A.L.; Bui, B.-H.T.; Vu, T.T.; Nguyen, H.-L.T.; Phung, B.-K.; Nguyen, V.-M.; Pham, V.-A.; Cao, V.-H.; Phan, T.-N. Leigh Syndrome T8993C Mitochondrial DNA Mutation: Heteroplasmy and the First Clinical Presentation in a Vietnamese Family. Mol. Med. Rep. 2018, 17, 6919–6925. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, S.; Lewis, B.; Mock, D.M.; Said, H.M.; Tarailo-Graovac, M.; Mattman, A.; van Karnebeek, C.D.; Thorburn, D.R.; Rodenburg, R.J.; Christodoulou, J. Leigh-Like Syndrome Due to Homoplasmic m.8993T>G Variant with Hypocitrullinemia and Unusual Biochemical Features Suggestive of Multiple Carboxylase Deficiency (MCD). JIMD Rep. 2016, 33, 99–107. [Google Scholar]
- Blok, R.B.; Gook, D.A.; Thorburn, D.R.; Dahl, H.H. Skewed Segregation of the MtDNA Nt 8993 (T-->G) Mutation in Human Oocytes. Am. J. Hum. Genet. 1997, 60, 1495–1501. [Google Scholar] [CrossRef] [Green Version]
- Burr, S.P.; Pezet, M.; Chinnery, P.F. Mitochondrial DNA Heteroplasmy and Purifying Selection in the Mammalian Female Germ Line. Dev. Growth Differ. 2018, 60, 21–32. [Google Scholar] [CrossRef] [Green Version]
- White, S.L.; Shanske, S.; McGill, J.J.; Mountain, H.; Geraghty, M.T.; DiMauro, S.; Dahl, H.H.; Thorburn, D.R. Mitochondrial DNA Mutations at Nucleotide 8993 Show a Lack of Tissue- or Age-Related Variation. J. Inherit. Metab. Dis. 1999, 22, 899–914. [Google Scholar] [CrossRef]
- Fassone, E.; Wedatilake, Y.; DeVile, C.J.; Chong, W.K.; Carr, L.J.; Rahman, S. Treatable Leigh-like Encephalopathy Presenting in Adolescence. BMJ Case Rep. 2013, 2013. [Google Scholar] [CrossRef]
- Licchetta, L.; Ferri, L.; Morgia, C.L.; Zenesini, C.; Caporali, L.; Valentino, M.L.; Minardi, R.; Fulitano, D.; Vito, L.D.; Mostacci, B.; et al. Epilepsy in MT-ATP6 - Related Mils/NARP: Correlation of Elettroclinical Features with Heteroplasmy. Ann. Clin. Transl. Neurol. 2021, 8, 704–710. [Google Scholar] [CrossRef]
- Distelmaier, F.; Huppke, P.; Pieperhoff, P.; Amunts, K.; Schaper, J.; Morava, E.; Ma yatepek, E.; Kohlhase, J.; Karenfort, M. Biotin-Responsive Basal Ganglia Disease: A Treatable Differential Diagnosis of Leigh Syndrome. JIMD Rep. 2014, 13, 53–57. [Google Scholar]
- Yatsuga, S.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T.; Kakuma, T.; Koga, Y. MELAS: A Nationwide Prospective Cohort Study of 96 Patients in Japan. Biochim. Biophys. Acta 2012, 1820, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Ricci, E.; Richard Koenigsberger, M.; Defendini, R.; Pavlakis, S.G.; DeVivo, D.C.; DiMauro, S.; Rowland, L.P. MELAS: An Original Case and Clinical Criteria for Diagnosis. Neuromuscul. Disord. 1992, 2, 125–135. [Google Scholar] [CrossRef]
- Kimata, K.G.; Gordan, L.; Ajax, E.T.; Davis, P.H.; Grabowski, T. A Case of Late-Onset MELAS. Arch. Neurol. 1998, 55, 722. [Google Scholar] [CrossRef] [Green Version]
- Sinnecker, T.; Andelova, M.; Mayr, M.; Rüegg, S.; Sinnreich, M.; Hench, J.; Frank, S.; Schaller, A.; Stippich, C.; Wuerfel, J.; et al. Diagnosis of Adult-Onset MELAS Syndrome in a 63-Year-Old Patient with Suspected Recurrent Strokes—A Case Report. BMC Neurol. 2019, 19, 91. [Google Scholar] [CrossRef] [Green Version]
- Parsons, T.; Weimer, L.; Engelstad, K.; Linker, A.; Battista, V.; Wei, Y.; Hirano, M.; DiMauro, S. Autonomic Symptoms in Carriers of the m.3243AϾG Mitochondrial DNA Mutation. Arch. Neurol. 2010, 67, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R. The Wide Phenotypic Spectrum of the Common MELAS-Causing Mutation. Neurol. Today 2007, 7, 26. [Google Scholar] [CrossRef]
- Liang, C.; Ahmad, K.; Sue, C.M. The Broadening Spectrum of Mitochondrial Disease: Shifts in the Diagnostic Paradigm. Biochim. Biophys. Acta 2014, 1840, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Kulikova, R.; Oskoui, M.; Sproule, D.M.; Battista, V.; Koenigsberger, D.Y.; Pascual, J.M.; Shanske, S.; et al. Natural History of MELAS Associated with Mitochondrial DNA m.3243A>G Genotype. Neurology 2011, 77, 1965–1971. [Google Scholar] [CrossRef] [Green Version]
- Weiduschat, N.; Kaufmann, P.; Mao, X.; Engelstad, K.M.; Hinton, V.; DiMauro, S.; Vivo, D.D.; Shungu, D. Cerebral Metabolic Abnormalities in A3243G Mitochondrial DNA Mutation Carriers. Neurology 2014, 82, 798–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J. Lifestyle Changes Normalize Serum Lactate Levels in an m.3243A>G Carrier. Am. J. Case Rep. 2021, 22, e930175. [Google Scholar] [CrossRef] [PubMed]
- Sofou, K.; de Coo, I.F.M.; Ostergaard, E.; Isohanni, P.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; Lönnqvist, T.; Bindoff, L.A.; et al. Phenotype-Genotype Correlations in Leigh Syndrome: New Insights from a Multicentre Study of 96 Patients. J. Med. Genet. 2018, 55, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Yiş, U.; Seneca, S.; Dırık, E.; Kurul, S.H.; Özer, E.; Çakmakçi, H.; De Meırleır, L. Unusual Findings in Leigh Syndrome Caused by T8993C Mutation. Eur. J. Paediatr. Neurol. 2009, 13, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.J.; Ríos, P.G.; Akman, H.O.; Sciacco, M.; De Vivo, D.C.; DiMauro, S. Long Survival in Patients with Leigh Syndrome and the m.10191T>C Mutation in MT-ND3: A Case Report and Review of the Literature. J. Child. Neurol. 2014, 29, NP105–NP110. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Lee, Y.-M.; Kim, S.M.; Han, S.Y.; Lee, J.B.; Han, S.-H. Ophthalmological Manifestations in Patients with Leigh Syndrome. Br. J. Ophthalmol. 2015, 99, 528–535. [Google Scholar] [CrossRef]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stöckler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; van den Heuvel, L. Mutations in the Complex I NDUFS2 Gene of Patients with Cardiomyopathy and Encephalomyopathy. Ann. Neurol. 2001, 49, 195–201. [Google Scholar] [CrossRef]
- Bénit, P.; Beugnot, R.; Chretien, D.; Giurgea, I.; De Lonlay-Debeney, P.; Issartel, J.-P.; Corral-Debrinski, M.; Kerscher, S.; Rustin, P.; Rötig, A.; et al. Mutant NDUFV2 Subunit of Mitochondrial Complex I Causes Early Onset Hypertrophic Cardiomyopathy and Encephalopathy. Hum. Mutat. 2003, 21, 582–586. [Google Scholar] [CrossRef]
- Tay, S.K.H.; Sacconi, S.; Akman, H.O.; Morales, J.F.; Morales, A.; De Vivo, D.C.; Shanske, S.; Bonilla, E.; DiMauro, S. Unusual Clinical Presentations in Four Cases of Leigh Disease, Cytochrome C Oxidase Deficiency, and SURF1 Gene Mutations. J. Child. Neurol. 2005, 20, 670–674. [Google Scholar] [CrossRef]
- Leslie, N.; Wang, X.; Peng, Y.; Valencia, C.A.; Khuchua, Z.; Hata, J.; Witte, D.; Huang, T.; Bove, K.E. Neonatal Multiorgan Failure Due to ACAD9 Mutation and Complex I Deficiency with Mitochondrial Hyperplasia in Liver, Cardiac Myocytes, Skeletal Muscle, and Renal Tubules. Hum. Pathol. 2016, 49, 27–32. [Google Scholar] [CrossRef]
- Finsterer, J. Rare Phenotypic Manifestations of MELAS. Yonsei Med. J. 2020, 61, 904–906. [Google Scholar] [CrossRef]
- Keilland, E.; Rupar, C.A.; Prasad, A.N.; Tay, K.Y.; Downie, A.; Prasad, C. The Expanding Phenotype of MELAS Caused by the m.3291T > C Mutation in the MT-TL1 Gene. Mol. Genet. Metab. Rep. 2016, 6, 64–69. [Google Scholar] [CrossRef]
- Mochizuki, H.; Joh, K.; Kawame, H.; Imadachi, A.; Nozaki, H.; Ohashi, T.; Usui, N.; Eto, Y.; Kanetsuna, Y.; Aizawa, S. Mitochondrial Encephalomyopathies Preceded by De-Toni-Debré-Fanconi Syndrome or Focal Segmental Glomerulosclerosis. Clin. Nephrol. 1996, 46, 347–352. [Google Scholar] [PubMed]
- Motoda, A.; Kurashige, T.; Sugiura, T.; Nakamura, T.; Yamawaki, T.; Arihiro, K.; Matsumoto, M. [A case of MELAS with G13513A mutation presenting with chronic kidney disease long before stroke-like episodes]. Rinsho Shinkeigaku 2013, 53, 446–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitun, S.; Massobrio, L.; Rubegni, A.; Nesti, C.; Castiglione Morelli, M.; Boccalini, S.; Galletto Pregliasco, A.; Budaj, I.; Deferrari, L.; Rosa, G.M.; et al. MELAS Syndrome with Cardiac Involvement: A Multimodality Imaging Approach. Case Rep. Cardiol. 2016, 2016, 1–4. [Google Scholar] [CrossRef]
- Strachan, J.; McLellan, A.; Kirkpatrick, M.; Hume, R.; Mechan, D. Ketoacidosis: An Unusual Presentation of MELAS. J. Inherit. Metab. Dis. 2001, 24, 409–410. [Google Scholar] [CrossRef]
- Chaig, M.R.; Zernotti, M.E.; Soria, N.W.; Romero, O.F.; Romero, M.F.; Gerez, N.M. A Mutation in Mitochondrial 12S RRNA, A827G, in Argentinean Family with Hearing Loss after Aminoglycoside Treatment. Biochem. Biophys. Res. Commun. 2008, 368, 631–636. [Google Scholar] [CrossRef]
- Dowlati, M.A.; Derakhshandeh-peykar, P.; Houshmand, M.; Farhadi, M.; Shojaei, A.; Fallah, M.; Mohammadi, E.; Tajdini, A.; Arastoo, S.; Tavakkoly-Bazzaz, J. Novel Nucleotide Changes in Mutational Analysis of Mitochondrial 12SrRNA Gene in Patients with Nonsyndromic and Aminoglycoside-Induced Hearing Loss. Mol. Biol. Rep. 2013, 40, 2689–2695. [Google Scholar] [CrossRef]
- Häkli, S.; Luotonen, M.; Sorri, M.; Majamaa, K. Mutations in the Two Ribosomal RNA Genes in Mitochondrial DNA among Finnish Children with Hearing Impairment. BMC Med. Genet. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheu-Grau, D.; Pérez-Delgado, L.; Gómez-Díaz, C.; Fraile-Rodrigo, J.; Montoya, J.; Ruiz-Pesini, E. Mitochondrial Ribosome and Ménière’s Disease: A Pilot Study. Eur. Arch. Otorhinolaryngol. 2012, 269, 2003–2008. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial DNA Variation in Human Radiation and Disease. Cell 2015, 163, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Manwaring, N.; Jones, M.M.; Wang, J.J.; Rochtchina, E.; Howard, C.; Mitchell, P.; Sue, C.M. Population Prevalence of the MELAS A3243G Mutation. Mitochondrion 2007, 7, 230–233. [Google Scholar] [CrossRef]
- Pierron, D.; Rocher, C.; Amati-Bonneau, P.; Reynier, P.; Martin-Négrier, M.-L.; Allouche, S.; Batandier, C.; de Camaret, B.M.; Godinot, C.; Rotig, A.; et al. New Evidence of a Mitochondrial Genetic Background Paradox: Impact of the J Haplogroup on the A3243G Mutation. BMC Med. Genet. 2008, 9, 41. [Google Scholar] [CrossRef]
- Delgado-Sánchez, R.; Zárate-Moysen, A.; Monsalvo-Reyes, A.; Herrero, M.D.; Ruiz-Pesini, E.; López-Pérez, M.; Montoya, J.; Montiel-Sosa, J.F. Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like Episodes (MELAS) with the A3243G Mutation of the TRNALeu (UUR) Gene of MtDNA in Native American Haplogroup B2. Rev. Neurol. 2007, 44, 18–22. [Google Scholar]
- Liu, G.; Shen, X.; Sun, Y.; Lv, Q.; Li, Y.; Du, A. Heteroplasmy and Phenotype Spectrum of the Mitochondrial TRNALeu (UUR) Gene m.3243A>G Mutation in Seven Han Chinese Families. J. Neurol. Sci. 2020, 408, 116562. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, S.; Govindaraj, P.; Sankaran, B.P.; Nagappa, M.; Kabekkodu, S.P.; Jayaram, P.; Mallya, S.; Deepha, S.; Ponmalar, J.N.J.; Arivinda, H.R.; et al. Contribution of Nuclear and Mitochondrial Gene Mutations in Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes (MELAS) Syndrome. J. Neurol. 2021, 268, 2192–2207. [Google Scholar] [CrossRef] [PubMed]
- Habbane, M.; Llobet, L.; Bayona-Bafaluy, M.P.; Bárcena, J.E.; Ceberio, L.; Gómez-Díaz, C.; Gort, L.; Artuch, R.; Montoya, J.; Ruiz-Pesini, E. Leigh Syndrome in a Pedigree Harboring the m.1555A>G Mutation in the Mitochondrial 12S RRNA. Genes 2020, 11, 1007. [Google Scholar] [CrossRef]
- Prasad, H.V.R.; Neelima, K.; Shalini, M.; Chandak, G.R. Leigh’s Syndrome: A Case Report. Internet J. Pediatr. Neonatol. 2006, 7, 1–9. [Google Scholar]
- Chen, Z.; Zhao, Z.; Ye, Q.; Chen, Y.; Pan, X.; Sun, B.; Huang, H.; Zheng, A. Mild Clinical Manifestation and Unusual Recovery upon Coenzyme Q10 Treatment in the First Chinese Leigh Syndrome Pedigree with Mutation m.10197 G>A. Mol. Med. Rep. 2015, 11, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.-D.; Yang, Y.-L.; Tang, N.L.S.; Kong, Q.-P.; Wu, S.-F.; Zhang, Y.-P. Mitochondrial DNA Haplogroup Y Is Associated to Leigh Syndrome in Chinese Population. Gene 2013, 512, 460–463. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.M.; Manfredi, G. Mitochondrial DNA Background Modifies the Bioenergetics of NARP/MILS ATP6 Mutant Cells. Hum. Mol. Genet. 2010, 19, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Angural, A.; Sharma, I.; Pandoh, P.; Sharma, V.; Spolia, A.; Rai, E.; Singh, V.; Razdan, S.; Pandita, K.K.; Sharma, S. A Case Report on a Novel MT-ATP6 Gene Variation in Atypical Mitochondrial Leigh Syndrome Associated with Bilateral Basal Ganglia Calcifications. Mitochondrion 2019, 46, 209–213. [Google Scholar] [CrossRef]
- Hong, C.-M.; Na, J.-H.; Park, S.; Lee, Y.-M. Clinical Characteristics of Early-Onset and Late-Onset Leigh Syndrome. Front. Neurol. 2020, 11, 267. [Google Scholar] [CrossRef]
- Tapias, V.; Mastroberardino, P.G.; Maio, R.D. Mitochondrial Dysfunction and Neurodegeneration; Frontiers Media SA: Lausanne, Switzerland, 2020. [Google Scholar]
- Wang, Y.; Brinton, R.D. Triad of Risk for Late Onset Alzheimer’s: Mitochondrial Haplotype, APOE Genotype and Chromosomal Sex. Front. Aging Neurosci. 2016, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Nalls, M.; Evans, J.R.; Breen, D.P.; Winder-Rhodes, S.; Morrison, K.E.; Morris, H.R.; Williams-Gray, C.H.; Barker, R.A.; Singleton, A.B.; et al. Two-Stage Association Study and Meta-Analysis of Mitochondrial DNA Variants in Parkinson Disease. Neurology 2013, 80, 2042–2048. [Google Scholar] [CrossRef]
- Arning, L.; Haghikia, A.; Taherzadeh-Fard, E.; Saft, C.; Andrich, J.; Pula, B.; Höxtermann, S.; Wieczorek, S.; Akkad, D.A.; Perrech, M.; et al. Mitochondrial Haplogroup H Correlates with ATP Levels and Age at Onset in Huntington Disease. J. Mol. Med. 2010, 88, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Conforti, F.L.; Rocchi, A.; Tessitore, A.; Muglia, M.; Tedeschi, G.; Panza, D.; Monsurrò, M.; Sola, P.; Mandrioli, J.; et al. Could Mitochondrial Haplogroups Play a Role in Sporadic Amyotrophic Lateral Sclerosis? Neurosci. Lett. 2004, 371, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Ghabaee, M.; Omranisikaroudi, M.; Amrisaroukolaei, S.; Meysamie, A.; Sahraian, M.A.; Bayati, A.; Sanati, M.H.; Houshman, M.; Sadeghian, H.; Vajihazaman, K. Mitochondrial Mutation in Iranian Patients with Multiple Sclerosis, Correlation between Haplogroups H, A and Clinical Manifestations. Cell. Mol. Neurobiol. 2009, 29, 341–346. [Google Scholar] [CrossRef]
- Serrano-Teruel, M.E.; Garcia-Vieites, M.; Rego-Perez, I.; Domenech-Garcia, N.; Blanco-Garcia, F.; Cuenca-Castillo, J.J.; Bautista-Hernandez, V. Mitochondrial DNA Haplogroups Influence the Risk of Aortic Stenosis. Asian Cardiovasc. Thorac. Ann. 2019, 27, 5–10. [Google Scholar] [CrossRef]
- Alwehaidah, M.S.; Bakhiet, M.; AlFadhli, S. Mitochondrial Haplogroup Reveals the Genetic Basis of Diabetes Mellitus Type 2 Comorbidity in Psoriasis. Med. Princ. Pract. 2021, 30, 62–68. [Google Scholar] [PubMed]
- Soto-Hermida, A.; Fernández-Moreno, M.; Pértega-Díaz, S.; Oreiro, N.; Fernández-López, C.; Blanco, F.J.; Rego-Pérez, I. Mitochondrial DNA Haplogroups Modulate the Radiographic Progression of Spanish Patients with Osteoarthritis. Rheumatol. Int. 2015, 35, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, R.G.; Fariña, F.O.; Rey, A.M.; Rego-Pérez, I.; García, F.J.B.; García, J.L.F. Relationship between the Dynamics of Telomere Loss in Peripheral Blood Leukocytes from Osteoarthritis Patients and Mitochondrial DNA Haplogroup. J. Rheumatol. 2021, jrheum.201316, (Online ahead of print). [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Mitochondriopathies | MELAS | Leigh Disease |
|---|---|---|
| Mutation | m.3243A>G | m.8993T>G |
| Gene | RNA Gene MT-TL1 | Protein Coding gene MT-ATP6 |
| Codon number | − | 156 |
| Amino acid change | tRNA Leu | Leu-Arg |
| Mitomap | Confirmed-Pathogenic | Confirmed-Pathogenic |
| MitoTIP | Pathogenic | − |
| Mamit-tRNA | Pathogenic | − |
| UniProt ID | − | P00846 |
| Polyphen Prediction | − | probably damaging |
| PANTHER | − | probably damaging |
| PROVEAN | − | Deleterious (−5.180) |
| Probands | Haplogroup | Variants | Locus | Nucleotide Changes | A.A Changes | PhyloTree Build 17 in HG Branch | Mitomaster Frequencies in HG Branch |
|---|---|---|---|---|---|---|---|
| P1 | U2e | 73 | MT-CR | A>G | CR | Reported | 96.46% |
| (MELAS) | 152 | MT-CR | T>C | CR | Reported | 90.26% | |
| 263 | MT-CR | A>G | CR | Reported | 96.46% | ||
| 508 | MT-CR | A>G | CR | Reported | 94.39% | ||
| 739 | MT-RNR1 | C>T | rRNA | Not reported | 0.29% | ||
| 750 | MT-RNR1 | A>G | rRNA | Reported | 100.00% | ||
| 1438 | MT-RNR1 | A>G | rRNA | Reported | 98.82% | ||
| 1811 | MT-RNR2 | A>G | rRNA | Reported | 94.39% | ||
| 2706 | MT-RNR2 | A>G | rRNA | Reported | 99.11% | ||
| 3116 | MT-RNR2 | C>T | rRNA | Not reported | 22.71% | ||
| 3720 | MT-ND1 | A>G | Q138Q | Reported | 98.23% | ||
| 4769 | MT-ND2 | A>G | M100M | Reported | 98.52% | ||
| 5390 | MT-ND2 | A>G | M307M | Reported | 98.52% | ||
| 5426 | MT-ND3 | T>C | H319H | Reported | 98.52% | ||
| 6045 | MT-COI | C>T | L48L | Reported | 98.52% | ||
| 6152 | MT-COI | T>C | V83V | Reported | 98.23% | ||
| 7028 | MT-COI | C>T | A375A | Reported | 98.80% | ||
| 8860 | MT-ATP6 | A>G | T112A | Reported | 99.41% | ||
| 10,876 | MT-ND4 | A>G | L39L | Reported | 99.11% | ||
| 11,197 | MT-ND4 | C>T | G146G | Not reported | 22.42% | ||
| 11,467 | MT-ND4 | A>G | L236L | Reported | 98.82% | ||
| 11,719 | MT-ND4 | G>A | G320G | Reported | 99.40% | ||
| 12,308 | MT-TL2 | A>G | tRNA | Reported | 98.80% | ||
| 12,372 | MT-ND5 | G>A | L12L | Reported | 99.70% | ||
| 13,020 | MT-ND5 | T>C | G228G | Reported | 99.11% | ||
| 13,359 | MT-ND5 | G>A | M341M | Not reported | 0.00% | ||
| 14,766 | MT-CYB | C>T | T7I | Reported | 99.70% | ||
| 15,326 | MT-CYB | A>G | T194A | Reported | 99.70% | ||
| 15,907 | MT-TT | A>G | tRNA | Reported | 98.82% | ||
| 16,051 | MT-CR | A>G | CR | Reported | 96.75% | ||
| 16,129 | MT-CR | G>C | CR | Reported | 95.28% | ||
| 16,183 | MT-CR | A>C | CR | Not considered | 75.52% | ||
| 16,189 | MT-CR | T>C | CR | Reported | 84.36% | ||
| P2 | H15 | 55 | MT-CR | T>C | CR | Reported | 63.33% |
| (MELAS) | 56 | MT-CR | insC | CR | Not reported | 6.67% | |
| 143 | MT-CR | G>A | CR | Not reported | 0.00% | ||
| 263 | MT-RNR1 | A>G | CR | Reported | 80.00% | ||
| 750 | MT-RNR1 | A>G | rRNA | Reported | 100.00% | ||
| 1438 | MT-RNR2 | A>G | rRNA | Reported | 96.70% | ||
| 2706 | MT-ND1 | A>G | rRNA | Reported | 95.60% | ||
| 3847 | MT-ND2 | T>C | L181L | Reported | 96.67% | ||
| 4769 | MT-COI | A>G | M100M | Reported | 96.70% | ||
| 6253 | MT-COI | T>C | M117T | Reported | 96.67% | ||
| 7028 | MT-ATP6 | C>T | A375A | Reported | 97.80% | ||
| 8860 | MT-CYB | A>G | T112A | Reported | 96.67% | ||
| 15,326 | MT-CR | A>G | T194A | Reported | 96.70% | ||
| P3 | H34 | 152 | MT-CR | T>C | CR | Not reported | 90.91% |
| (LS) | 263 | MT-CR | A>G | CR | Reported | 100.00% | |
| 315 | MT-CR | insC | CR | Not reported | 45.46% | ||
| 508 | MT-RNR1 | A>G | CR | Not reported | 0.00% | ||
| 750 | MT-RNR1 | A>G | rRNA | Reported | 100.00% | ||
| 1438 | MT-RNR1 | A>G | rRNA | Reported | 100.00% | ||
| 4769 | MT-ND2 | A>G | M100M | Reported | 100.00% | ||
| 8860 | MT-ATP6 | A>G | T112A | Reported | 100.00% | ||
| 15,326 | MT-CYB | A>G | T194A | Reported | 100.00% | ||
| 15,519 | MT-CYB | T>C | L258P | Reported | 100.00% | ||
| 15,948 | MT-TT | A>G | tRNA | Not reported | 0.00% | ||
| 16,093 | MT-CR | T>C | CR | Not reported | 45.46% | ||
| 16,291 | MT-CR | C>T | CR | Reported | 90.91% | ||
| 16,519 | MT-CR | T>C | CR | Reported | 100.00% |
| Clinical Evaluation | MELAS | Leigh Disease | |
|---|---|---|---|
| Proband | P1 | P2 | P3 |
| Gender | Male | Male | Female |
| Age at onset of the disease | 14 years old | 12 years old | Few months after birth |
| Duration of the disease | 1 month | 9 years | 4 years |
| Family history of MEMP | − | – | – |
| Epileptic seizures | + | + | + |
| Psychosis | + | + | − |
| Psychomotor retardation | – | − | + |
| Confusion | + | − | − |
| Behaviour changes | + | + | + |
| Dementia | − | + | − |
| Episodes like stroke | − | − | − |
| Headache | + | − | − |
| Eye deviation during seizures | − | − | + |
| Speech delay | − | − | + |
| Hemiparesis | + | + | − |
| Muscle weakness | + | + | + |
| Muscle twitches | + | − | + |
| Associated vomiting | + | + | − |
| Preceding infection | − | + | + |
| Lactate acidosis | + | + | + |
| MRI changes | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawod, P.G.A.; Jancic, J.; Marjanovic, A.; Brankovic, M.; Jankovic, M.; Samardzic, J.; Gamil Anwar Dawod, A.; Novakovic, I.; Abdel Motaleb, F.I.; Radlovic, V.; et al. Mutational Analysis and mtDNA Haplogroup Characterization in Three Serbian Cases of Mitochondrial Encephalomyopathies and Literature Review. Diagnostics 2021, 11, 1969. https://doi.org/10.3390/diagnostics11111969
Dawod PGA, Jancic J, Marjanovic A, Brankovic M, Jankovic M, Samardzic J, Gamil Anwar Dawod A, Novakovic I, Abdel Motaleb FI, Radlovic V, et al. Mutational Analysis and mtDNA Haplogroup Characterization in Three Serbian Cases of Mitochondrial Encephalomyopathies and Literature Review. Diagnostics. 2021; 11(11):1969. https://doi.org/10.3390/diagnostics11111969
Chicago/Turabian StyleDawod, Phepy G. A., Jasna Jancic, Ana Marjanovic, Marija Brankovic, Milena Jankovic, Janko Samardzic, Ayman Gamil Anwar Dawod, Ivana Novakovic, Fayda I. Abdel Motaleb, Vladimir Radlovic, and et al. 2021. "Mutational Analysis and mtDNA Haplogroup Characterization in Three Serbian Cases of Mitochondrial Encephalomyopathies and Literature Review" Diagnostics 11, no. 11: 1969. https://doi.org/10.3390/diagnostics11111969
APA StyleDawod, P. G. A., Jancic, J., Marjanovic, A., Brankovic, M., Jankovic, M., Samardzic, J., Gamil Anwar Dawod, A., Novakovic, I., Abdel Motaleb, F. I., Radlovic, V., Kostic, V. S., & Nikolic, D. (2021). Mutational Analysis and mtDNA Haplogroup Characterization in Three Serbian Cases of Mitochondrial Encephalomyopathies and Literature Review. Diagnostics, 11(11), 1969. https://doi.org/10.3390/diagnostics11111969