
Development of Quantitative Rapid Isothermal Amplification Assay for Leishmania donovani

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Theoretical Considerations
2.2. Sample Source
2.3. DNA Standard Preparation
2.4. RPA and Real-Time qPCR Assays
2.5. Curation of Q-RPA Amplification Data for Detection and Quantification
2.6. Statistical Analysis
3. Results
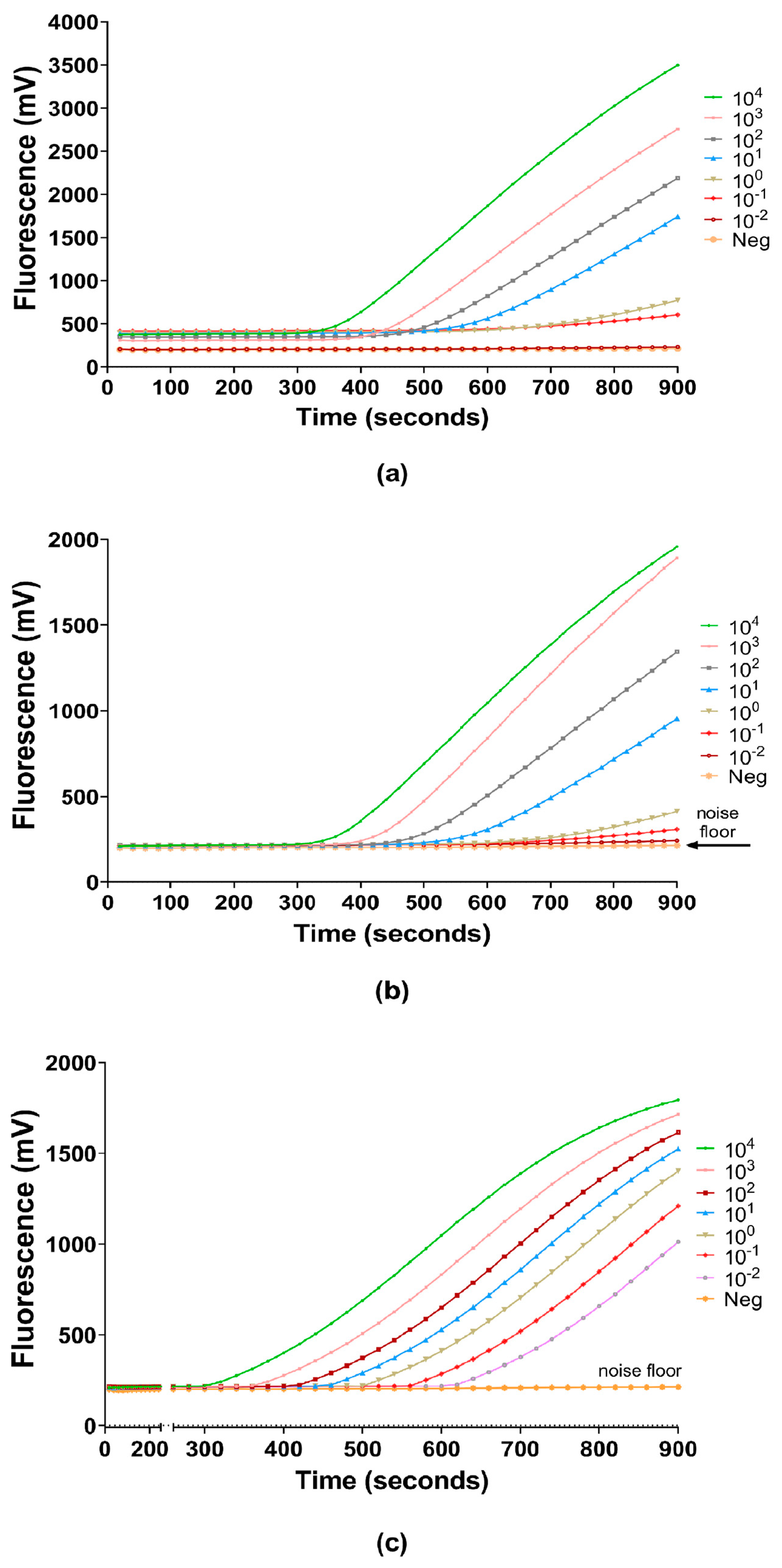
3.1. Generalization, Linearity and Reproducibility of Q-RPA Amplification
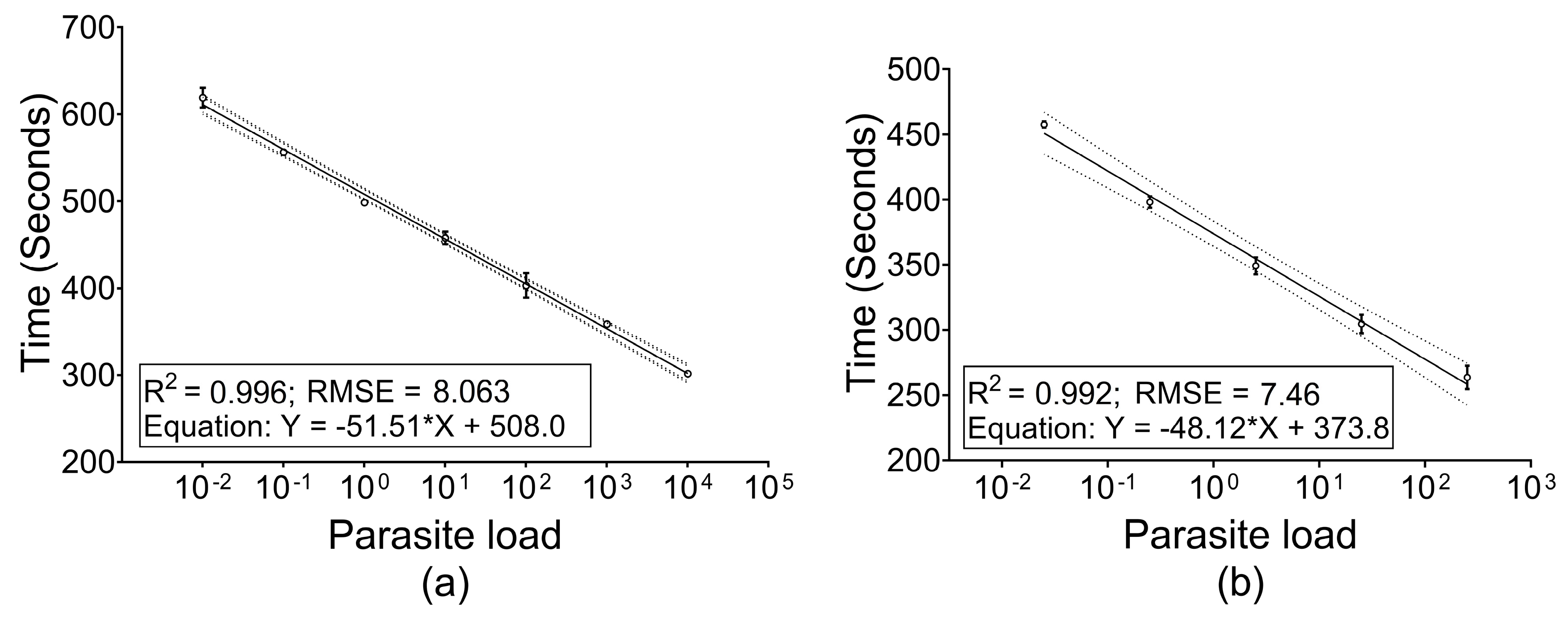
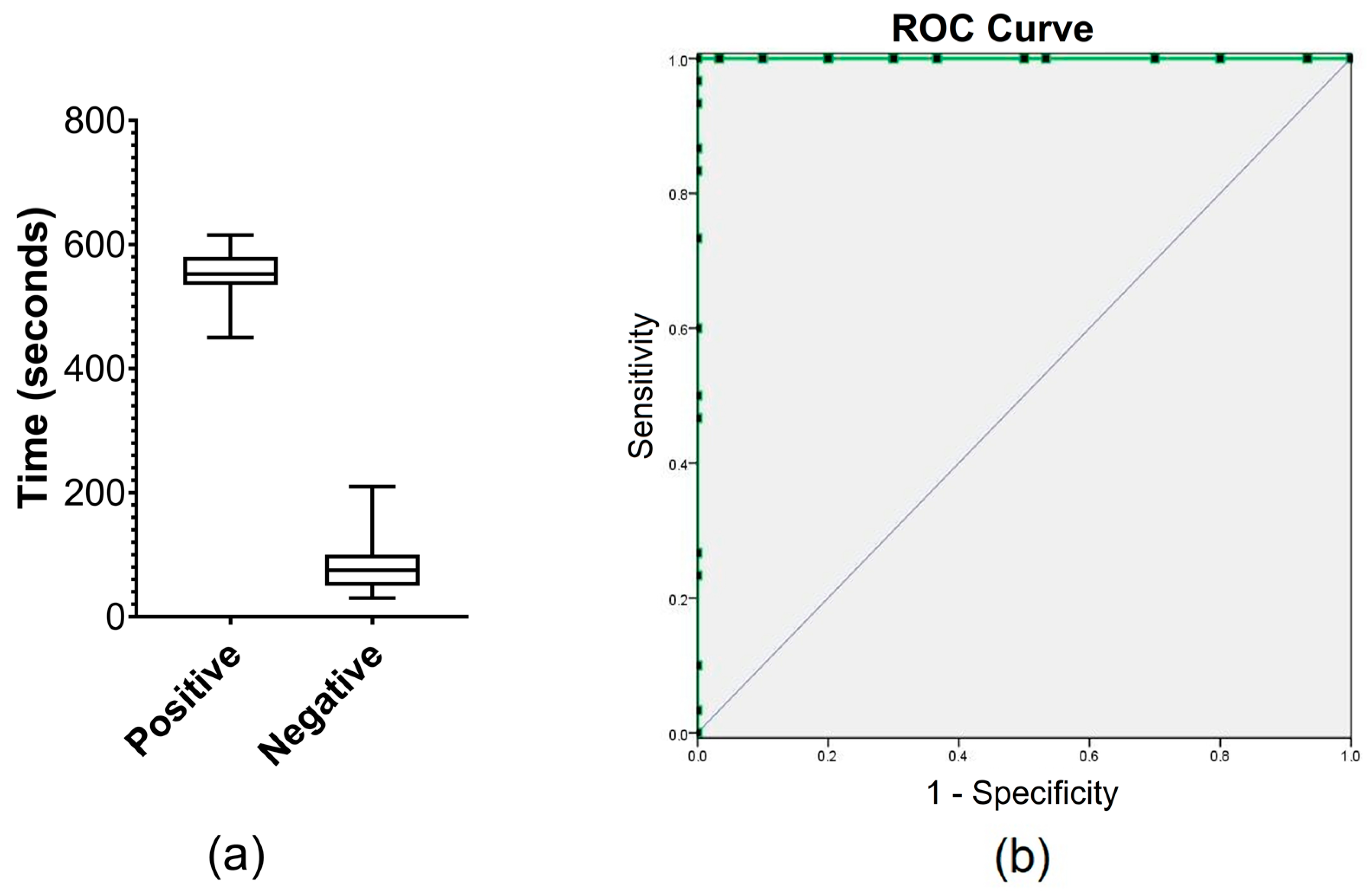
3.2. Threshold E-Phase and Limit of Detection (LOD)
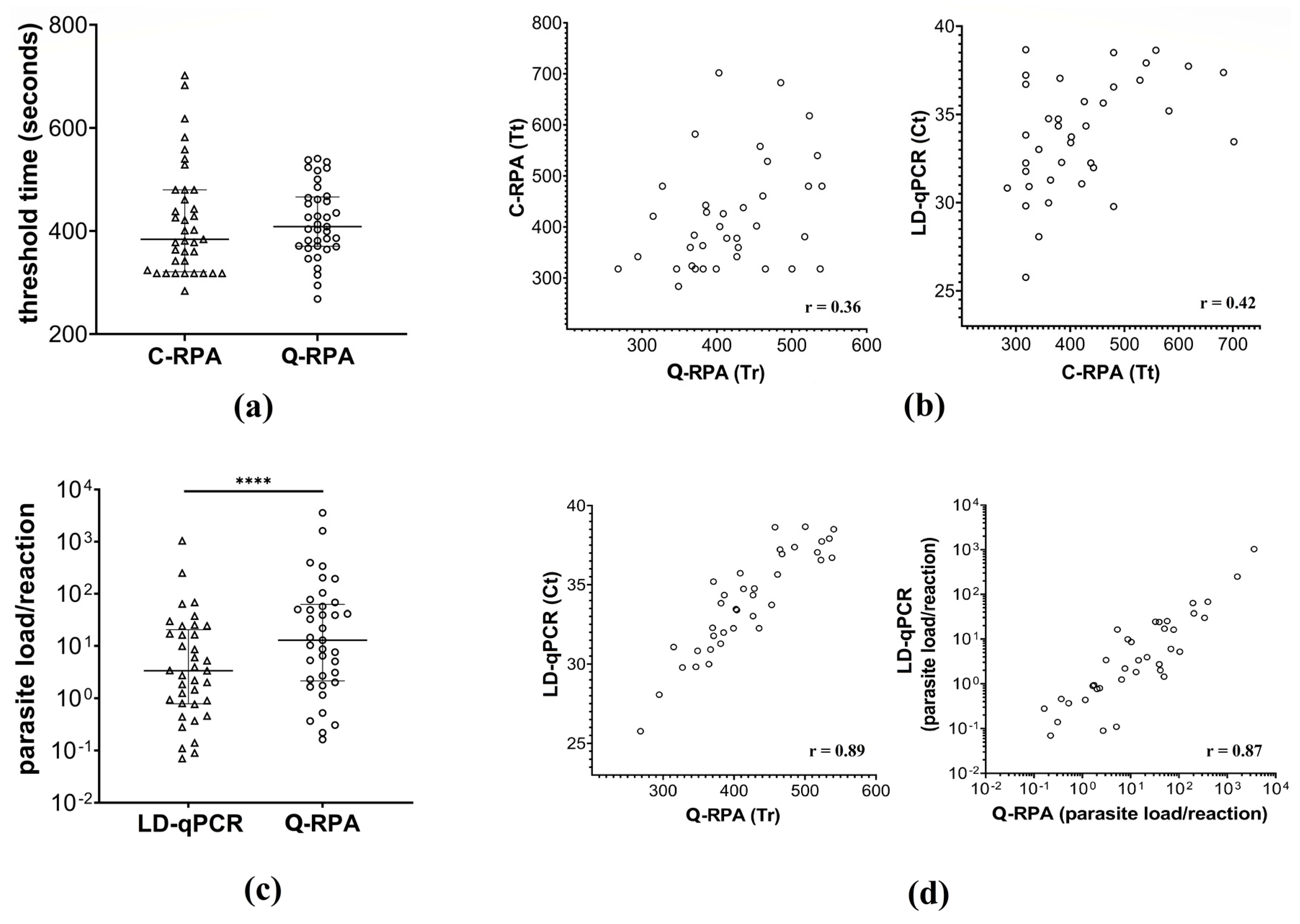
3.3. Q-RPA Quantification and Comparison with C-RPA and LD-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Carman, B. Molecular techniques should now replace cell culture in diagnostic virology laboratories. Rev. Med. Virol. 2001, 11, 347–349. [Google Scholar] [CrossRef]
- Limaye, A.P.; Huang, M.-L.; Leisenring, W.; Stensland, L.; Corey, L.; Boeckh, M. Cytomegalovirus (CMV) DNA Load in Plasma for the Diagnosis of CMV Disease before Engraftment in Hematopoietic Stem-Cell Transplant Recipients. J. Infect. Dis. 2001, 183, 377–382. [Google Scholar] [CrossRef]
- Cesaro, S.; Murrone, A.; Mengoli, C.; Pillon, M.; Biasolo, M.A.; Calore, E.; Tridello, G.; Varotto, S.; Alaggio, R.; Zanesco, L.; et al. The real-time polymerase chain reaction-guided modulation of immunosuppression enables the pre-emptive management of Epstein–Barr virus reactivation after allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2005, 128, 224–233. [Google Scholar] [CrossRef]
- Beshir, K.B.; Hallett, R.L.; Eziefula, A.C.; Bailey, R.; Watson, J.; Wright, S.G.; Chiodini, P.L.; Polley, S.D.; Sutherland, C.J. Measuring the efficacy of anti-malarial drugs in vivo: Quantitative PCR measurement of parasite clearance. Malar. J. 2010, 9, 312. [Google Scholar] [CrossRef]
- Pas, S.D.; Noppornpanth, S.; Eijk, A.A.v.d.; Man, R.A.d.; Niesters, H.G.M. Quantification of the newly detected lamivudine resistant YSDD variants of Hepatitis B virus using molecular beacons. J. Clin. Virol. 2005, 32, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Lion, T.; Baumgartinger, R.; Watzinger, F.; Matthes-Martin, S.; Suda, M.; Preuner, S.; Futterknecht, B.; Lawitschka, A.; Peters, C.; Pötschger, U.; et al. Molecular monitoring of adenovirus in peripheral blood after allogeneic bone marrow transplantation permits early diagnosis of disseminated disease. Blood 2003, 102, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, K.; Dandachi, N.; Trattner, M.; Vogl, G.; Doppelmayr, H.; Moré, E.; Staudach, A.; Dietze, O.; Hauser-Kronberger, C. Comparison of Real-Time PCR Signal-Amplified In Situ Hybridization and Conventional PCR for Detection and Quantification of Human Papillomavirus in Archival Cervical Cancer Tissue. J. Clin. Microbiol. 2004, 42, 3758–3765. [Google Scholar] [CrossRef]
- Snijders, P.J.; van den Brule, A.J.; Meijer, C.J. The clinical relevance of human papillomavirus testing: Relationship between analytical and clinical sensitivity. J. Pathol. 2003, 201, 1–6. [Google Scholar] [CrossRef]
- Watzinger, F.; Suda, M.; Preuner, S.; Baumgartinger, R.; Ebner, K.; Baskova, L.; Niesters, H.G.M.; Lawitschka, A.; Lion, T. Real-Time Quantitative PCR Assays for Detection and Monitoring of Pathogenic Human Viruses in Immunosuppressed Pediatric Patients. J. Clin. Microbiol. 2004, 42, 5189–5198. [Google Scholar] [CrossRef]
- Freeman, W.M.; Walker, S.J.; Vrana, K.E. Quantitative RT-PCR: Pitfalls and Potential. BioTechniques 1999, 26, 112–125. [Google Scholar] [CrossRef]
- Neuzil, P.; Zhang, C.; Pipper, J.; Oh, S.; Zhuo, L. Ultra fast miniaturized real-time PCR: 40 cycles in less than six minutes. Nucleic Acids Res. 2006, 34, e77. [Google Scholar] [CrossRef]
- Quan, P.-L.; Sauzade, M.; Brouzes, E. dPCR: A Technology Review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Xu, B.; Li, D. Miniature real time PCR on chip with multi-channel fiber optical fluorescence detection module. Biomed. Microdevices 2007, 9, 443–449. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, F.; Li, Q.; Wang, L.; Fan, C. Isothermal Amplification of Nucleic Acids. Chem. Rev. 2015, 115, 12491–12545. [Google Scholar] [CrossRef]
- Craw, P.; Balachandran, W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: A critical review. Lab Chip 2012, 12, 2469–2486. [Google Scholar] [CrossRef]
- Crannell, Z.A.; Rohrman, B.; Richards-Kortum, R. Quantification of HIV-1 DNA Using Real-Time Recombinase Polymerase Amplification. Anal. Chem. 2014, 86, 5615–5619. [Google Scholar] [CrossRef]
- Hsieh, K.; Patterson, A.S.; Ferguson, B.S.; Plaxco, K.W.; Soh, H.T. Rapid, Sensitive, and Quantitative Detection of Pathogenic DNA at the Point of Care through Microfluidic Electrochemical Quantitative Loop-Mediated Isothermal Amplification. Angew. Chem. Int. Ed. 2012, 51, 4896–4900. [Google Scholar] [CrossRef]
- Nixon, G.; Garson, J.A.; Grant, P.; Nastouli, E.; Foy, C.A.; Huggett, J.F. Comparative Study of Sensitivity, Linearity, and Resistance to Inhibition of Digital and Nondigital Polymerase Chain Reaction and Loop Mediated Isothermal Amplification Assays for Quantification of Human Cytomegalovirus. Anal. Chem. 2014, 86, 4387–4394. [Google Scholar] [CrossRef]
- Shen, F.; Davydova, E.K.; Du, W.; Kreutz, J.E.; Piepenburg, O.; Ismagilov, R.F. Digital Isothermal Quantification of Nucleic Acids via Simultaneous Chemical Initiation of Recombinase Polymerase Amplification Reactions on SlipChip. Anal. Chem. 2011, 83, 3533–3540. [Google Scholar] [CrossRef]
- van der Meide, W.F.; Schoone, G.J.; Faber, W.R.; Zeegelaar, J.E.; de Vries, H.J.C.; Özbel, Y.; Lai A Fat, R.F.M.; Coelho, L.I.A.R.C.; Kassi, M.; Schallig, H.D.F.H. Quantitative Nucleic Acid Sequence-Based Assay as a New Molecular Tool for Detection and Quantification of Leishmania Parasites in Skin Biopsy Samples. J. Clin. Microbiol. 2005, 43, 5560–5566. [Google Scholar] [CrossRef]
- Khan, M.A.A.; Chowdhury, R.; Nath, R.; Hansen, S.; Nath, P.; Maruf, S.; Abd El Wahed, A.; Mondal, D. Imported cutaneous leishmaniasis: Molecular investigation unveils Leishmania major in Bangladesh. Parasites Vectors 2019, 12, 527. [Google Scholar] [CrossRef]
- Maurício, I.L.; Stothard, J.R.; Miles, M.A. The Strange Case of Leishmania chagasi. Parasitol. Today 2000, 16, 188–189. [Google Scholar] [CrossRef]
- Siriwardana, H.V.Y.D.; Karunanayake, P.; Goonerathne, L.; Karunaweera, N.D. Emergence of visceral leishmaniasis in Sri Lanka: A newly established health threat. Pathog. Glob. Health 2017, 111, 317–326. [Google Scholar] [CrossRef]
- Mondal, D.; Ghosh, P.; Khan, M.A.A.; Hossain, F.; Böhlken-Fascher, S.; Matlashewski, G.; Kroeger, A.; Olliaro, P.; Abd El Wahed, A. Mobile suitcase laboratory for rapid detection of Leishmania donovani using recombinase polymerase amplification assay. Parasites Vectors 2016, 9, 281. [Google Scholar] [CrossRef]
- Nzelu, C.O.; Kato, H.; Peters, N.C. Loop-mediated isothermal amplification (LAMP): An advanced molecular point-of-care technique for the detection of Leishmania infection. PLoS Negl. Trop. Dis. 2019, 13, e0007698. [Google Scholar] [CrossRef]
- Sudarshan, M.; Weirather, J.L.; Wilson, M.E.; Sundar, S. Study of parasite kinetics with antileishmanial drugs using real-time quantitative PCR in Indian visceral leishmaniasis. J. Antimicrob. Chemother. 2011, 66, 1751–1755. [Google Scholar] [CrossRef]
- Mary, C.; Faraut, F.; Drogoul, M.P.; Xeridat, B.; Schleinitz, N.; Cuisenier, B.; Dumon, H. Reference values for Leishmania infantum parasitemia in different clinical presentations: Quantitative polymerase chain reaction for therapeutic monitoring and patient follow-up. Am. J. Trop. Med. Hyg. 2006, 75, 858–863. [Google Scholar] [CrossRef][Green Version]
- Bossolasco, S.; Gaiera, G.; Olchini, D.; Gulletta, M.; Martello, L.; Bestetti, A.; Bossi, L.; Germagnoli, L.; Lazzarin, A.; Uberti-Foppa, C.; et al. Real-Time PCR Assay for Clinical Management of Human Immunodeficiency Virus-Infected Patients with Visceral Leishmaniasis. J. Clin. Microbiol. 2003, 41, 5080–5084. [Google Scholar] [CrossRef]
- Mary, C.; Faraut, F.; Lascombe, L.; Dumon, H. Quantification of Leishmania infantum DNA by a Real-Time PCR Assay with High Sensitivity. J. Clin. Microbiol. 2004, 42, 5249–5255. [Google Scholar] [CrossRef]
- Hossain, F.; Ghosh, P.; Khan, M.A.A.; Duthie, M.S.; Vallur, A.C.; Picone, A.; Howard, R.F.; Reed, S.G.; Mondal, D. Real-time PCR in detection and quantitation of Leishmania donovani for the diagnosis of Visceral Leishmaniasis patients and the monitoring of their response to treatment. PLoS ONE 2017, 12, e0185606. [Google Scholar] [CrossRef]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2019, 144, 31–67. [Google Scholar] [CrossRef]
- Ghosh, P.; Sharma, A.; Bhattarai, N.R.; Abhishek, K.; Nisansala, T.; Kumar, A.; Böhlken-Fascher, S.; Chowdhury, R.; Khan, M.A.A.; Faisal, K.; et al. A Multi-Country, Single-Blinded, Phase 2 Study to Evaluate a Point-of-Need System for Rapid Detection of Leishmaniasis and Its Implementation in Endemic Settings. Microorganisms 2021, 9, 588. [Google Scholar] [CrossRef]
- Powell, M.L.; Bowler, F.R.; Martinez, A.J.; Greenwood, C.J.; Armes, N.; Piepenburg, O. New Fpg probe chemistry for direct detection of recombinase polymerase amplification on lateral flow strips. Anal. Biochem. 2018, 543, 108–115. [Google Scholar] [CrossRef]
- TwistDx. TwistAmp® Combined Instruction Manual; TwistDx: Cambridge, UK, 2014. [Google Scholar]
- Gordon, M.I.; Klemer, D.P.; Fuller, S.L.; Chang, J.H.; Klemer, D.R.; Putnam, M.L. Mathematical modeling of a real-time isothermal amplification assay for Erwinia amylovora. Eng. Rep. 2019, 1, e12047. [Google Scholar] [CrossRef]
- Moody, C.; Newell, H.; Viljoen, H. A mathematical model of recombinase polymerase amplification under continuously stirred conditions. Biochem. Eng. J. 2016, 112, 193–201. [Google Scholar] [CrossRef]
- Parikh, R.; Mathai, A.; Parikh, S.; Chandra Sekhar, G.; Thomas, R. Understanding and using sensitivity, specificity and predictive values. Indian J. Ophthalmol. 2008, 56, 45–50. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef]
- Kalsi, S.; Valiadi, M.; Tsaloglou, M.-N.; Parry-Jones, L.; Jacobs, A.; Watson, R.; Turner, C.; Amos, R.; Hadwen, B.; Buse, J.; et al. Rapid and sensitive detection of antibiotic resistance on a programmable digital microfluidic platform. Lab Chip 2015, 15, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Ceccarelli, M.; Diotallevi, A.; Menotta, M.; Magnani, M. Real-time PCR applications for diagnosis of leishmaniasis. Parasites Vectors 2018, 11, 273. [Google Scholar] [CrossRef]
- Kocher, A.; ValiÈRe, S.; BaÑUls, A.-L.; Murienne, J. High-throughput sequencing of kDNA amplicons for the analysis of Leishmania minicircles and identification of Neotropical species. Parasitology 2017, 145, 585–594. [Google Scholar] [CrossRef]
- Alonso, D.P.; Costa, D.L.; de Mendonça, I.L.; Costa, C.H.N.; Ribolla, P.E.M. Heterogeneity of Leishmania infantum chagasi Kinetoplast DNA in Teresina (Brazil). Am. Soc. Trop. Med. Hyg. 2010, 82, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; Jeronimo, S.M.B.; Gautam, S.; Sundar, S.; Kang, M.; Kurtz, M.A.; Haque, R.; Schriefer, A.; Talhari, S.; Carvalho, E.M.; et al. Serial Quantitative PCR Assay for Detection, Species Discrimination, and Quantification of Leishmania spp. in Human Samples. J. Clin. Microbiol. 2011, 49, 3892–3904. [Google Scholar] [CrossRef] [PubMed]
- Jara, M.; Adaui, V.; Valencia, B.M.; Martinez, D.; Alba, M.; Castrillon, C.; Cruz, M.; Cruz, I.; Van der Auwera, G.; Llanos-Cuentas, A.; et al. Real-Time PCR Assay for Detection and Quantification of Leishmania (viannia) Organisms in Skin and Mucosal Lesions: Exploratory Study of Parasite Load and Clinical Parameters. J. Clin. Microbiol. 2013, 51, 1826–1833. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inter-Assay Variation of Tr in Seconds (NFW) | Inter-Assay Variation of Tr in Seconds (CSB) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parasite load | Assay 1 | Assay 2 | Mean | SD | CV (%) | Parasite load | Assay 1 | Assay 2 | Assay 3 | Mean | SD | CV (%) |
| 1 × 104 | 301.32 | 302.02 | 301.67 | 0.50 | 0.17 | 2.5 × 102 | 262.67 | 255.33 | 273.10 | 263.70 | 8.93 | 3.39 |
| 1 × 103 | 360.45 | 357.02 | 358.73 | 2.43 | 0.68 | 2.5 × 101 | 304.77 | 297.56 | 311.60 | 304.64 | 7.02 | 2.30 |
| 1 × 102 | 413.19 | 393.14 | 403.16 | 14.18 | 3.52 | 2.5 × 100 | 341.96 | 354.85 | 350.90 | 349.24 | 6.60 | 1.89 |
| 1 × 101 | 452.70 | 462.87 | 457.79 | 7.19 | 1.57 | 2.5 × 10−1 | 395.85 | 403.17 | 395.08 | 398.03 | 4.46 | 1.12 |
| 1 × 100 | 496.73 | 500.12 | 498.42 | 2.40 | 0.48 | 2.5 × 10−2 | 459.90 | 455.29 | 457.57 | 457.58 | 2.30 | 0.50 |
| 1 × 10−1 | 555.79 | 556.92 | 556.36 | 0.80 | 0.14 | |||||||
| 1 × 10−2 | 610.96 | 626.92 | 618.94 | 11.29 | 1.82 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.A.A.; Faisal, K.; Chowdhury, R.; Ghosh, P.; Hossain, F.; Weidmann, M.; Mondal, D.; Abd El Wahed, A. Development of Quantitative Rapid Isothermal Amplification Assay for Leishmania donovani. Diagnostics 2021, 11, 1963. https://doi.org/10.3390/diagnostics11111963
Khan MAA, Faisal K, Chowdhury R, Ghosh P, Hossain F, Weidmann M, Mondal D, Abd El Wahed A. Development of Quantitative Rapid Isothermal Amplification Assay for Leishmania donovani. Diagnostics. 2021; 11(11):1963. https://doi.org/10.3390/diagnostics11111963
Chicago/Turabian StyleKhan, Md Anik Ashfaq, Khaledul Faisal, Rajashree Chowdhury, Prakash Ghosh, Faria Hossain, Manfred Weidmann, Dinesh Mondal, and Ahmed Abd El Wahed. 2021. "Development of Quantitative Rapid Isothermal Amplification Assay for Leishmania donovani" Diagnostics 11, no. 11: 1963. https://doi.org/10.3390/diagnostics11111963
APA StyleKhan, M. A. A., Faisal, K., Chowdhury, R., Ghosh, P., Hossain, F., Weidmann, M., Mondal, D., & Abd El Wahed, A. (2021). Development of Quantitative Rapid Isothermal Amplification Assay for Leishmania donovani. Diagnostics, 11(11), 1963. https://doi.org/10.3390/diagnostics11111963