
Comprehensive Molecular Analysis of DMD Gene Increases the Diagnostic Value of Dystrophinopathies: A Pilot Study in a Southern Italy Cohort of Patients

 , ,
, ,  , and
, and
Abstract
:1. Introduction
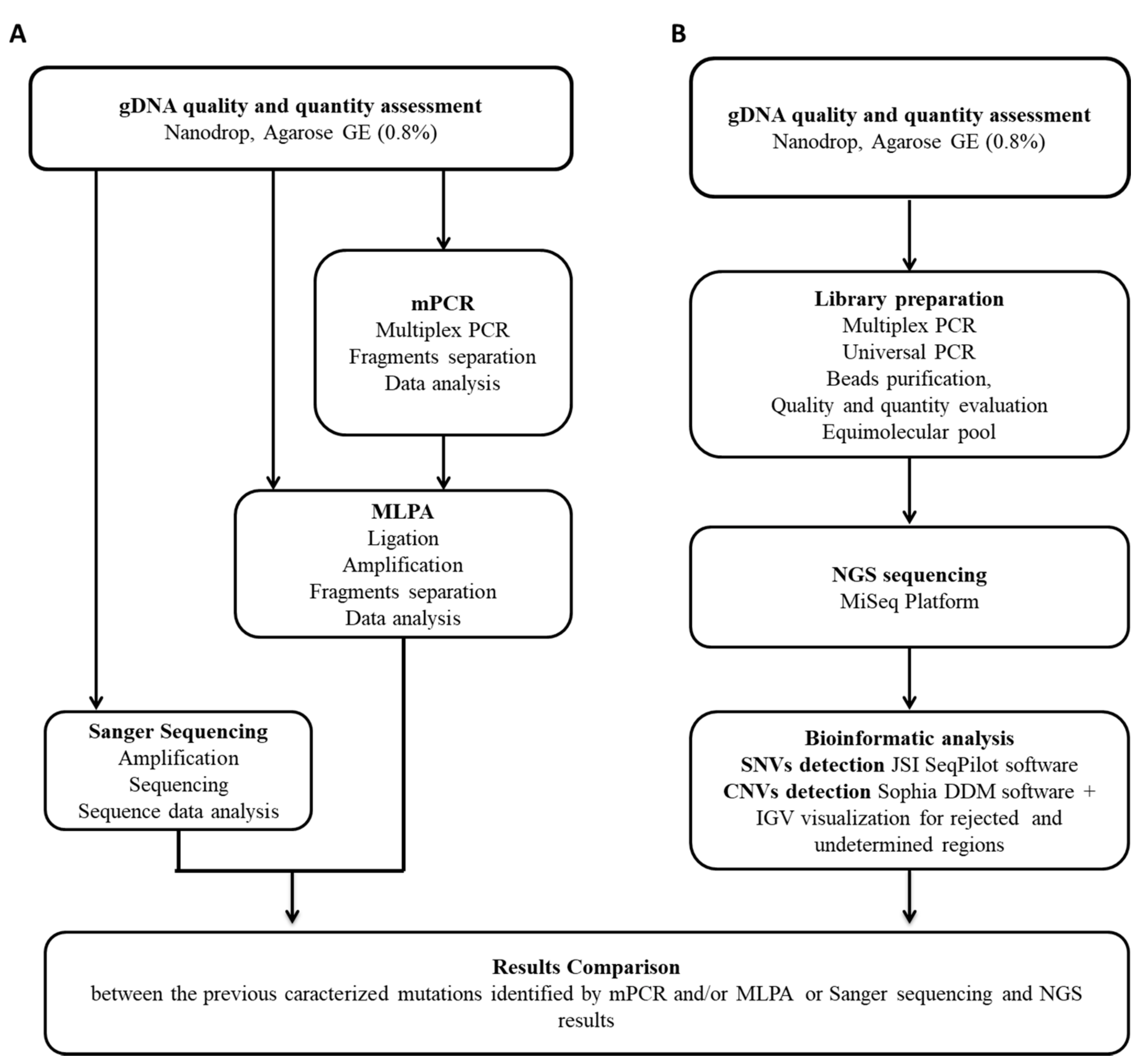
2. Materials and Methods
2.1. Study Population and DNA Samples
2.2. DMD Gene Traditional Molecular Analysis
2.3. NGS Analysis
2.4. NGS Sequence Data Analysis
3. Results
3.1. NGS Sequence Coverage
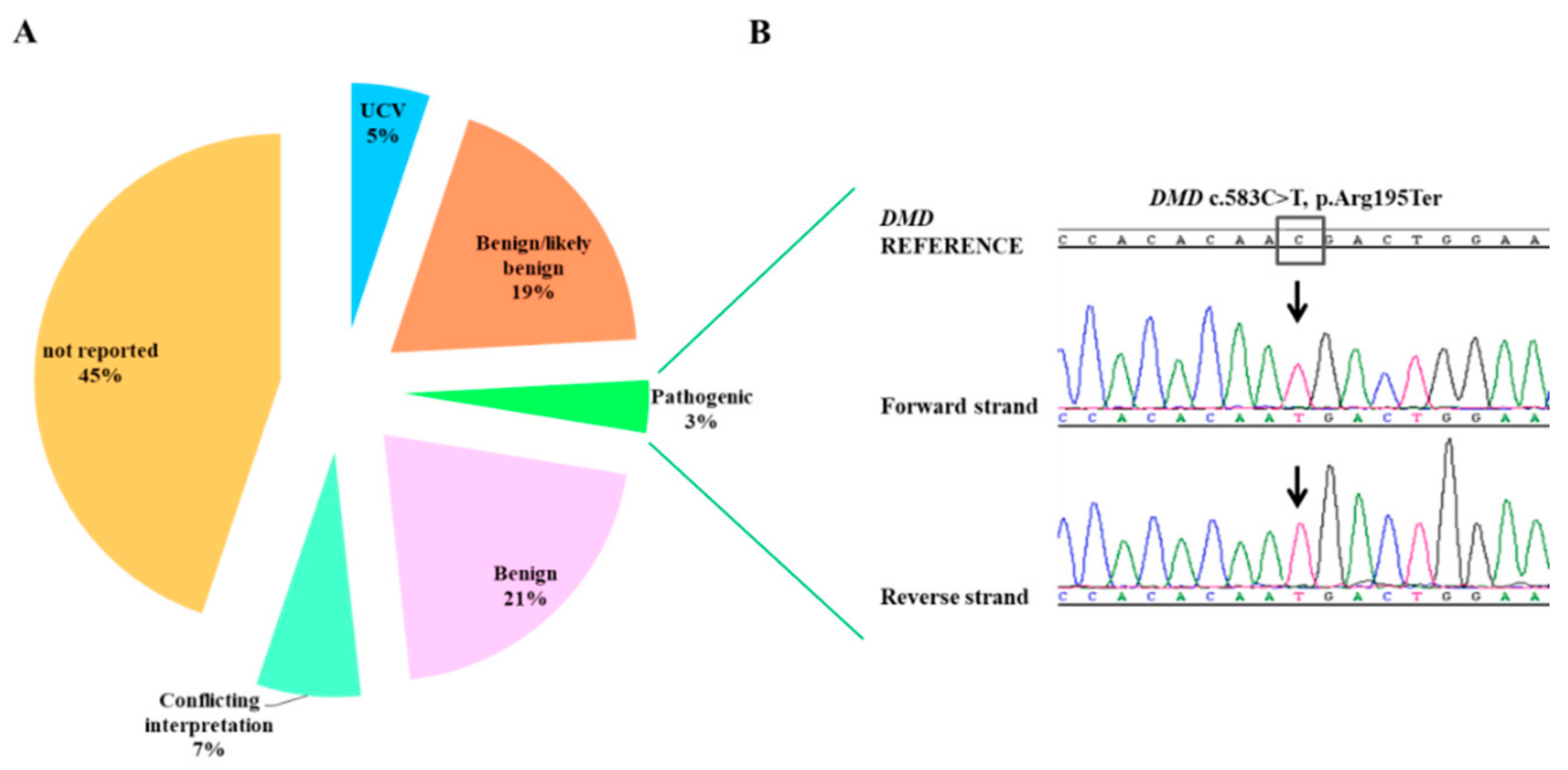
3.2. Point Mutation Identification
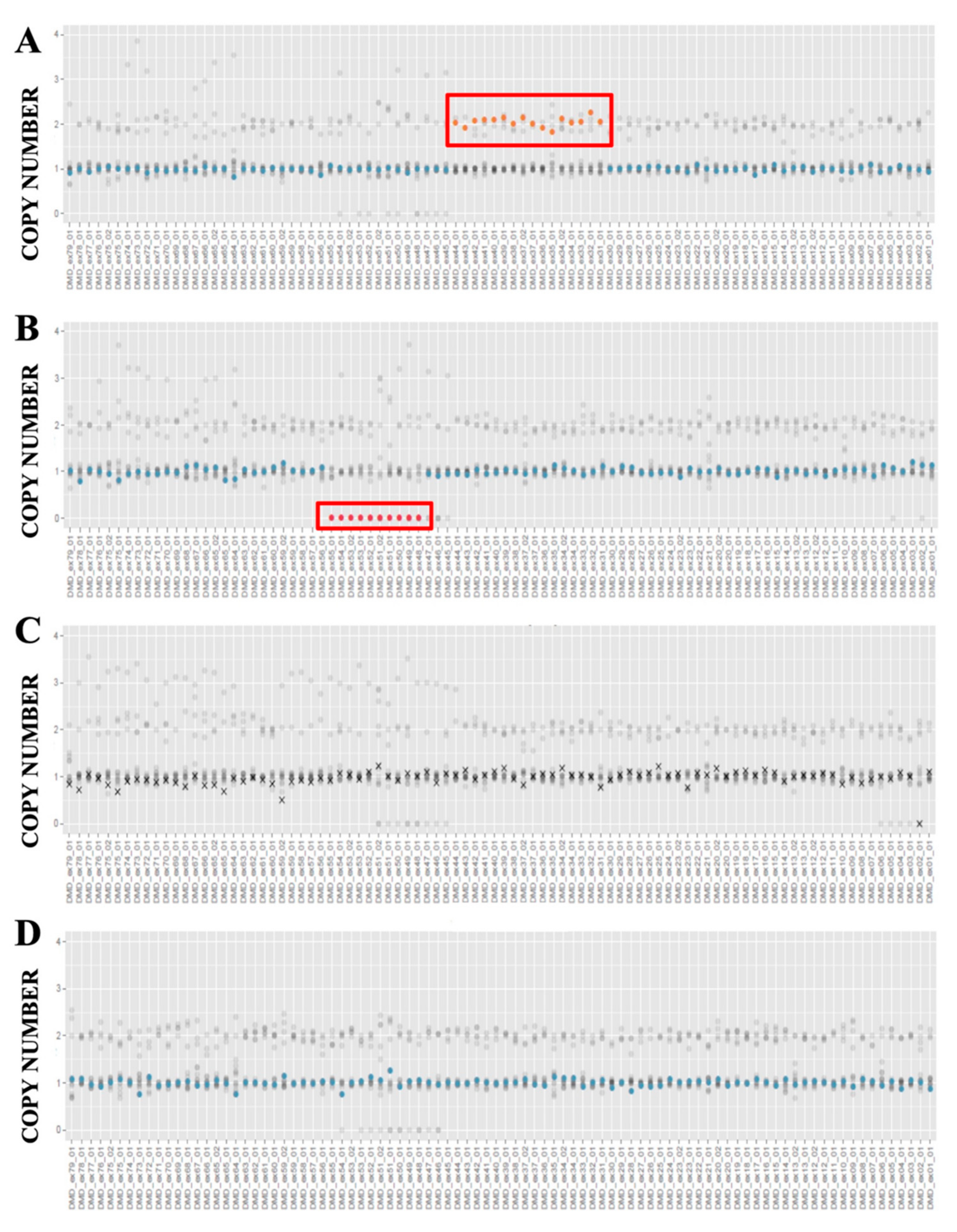
3.3. CNVs Detection
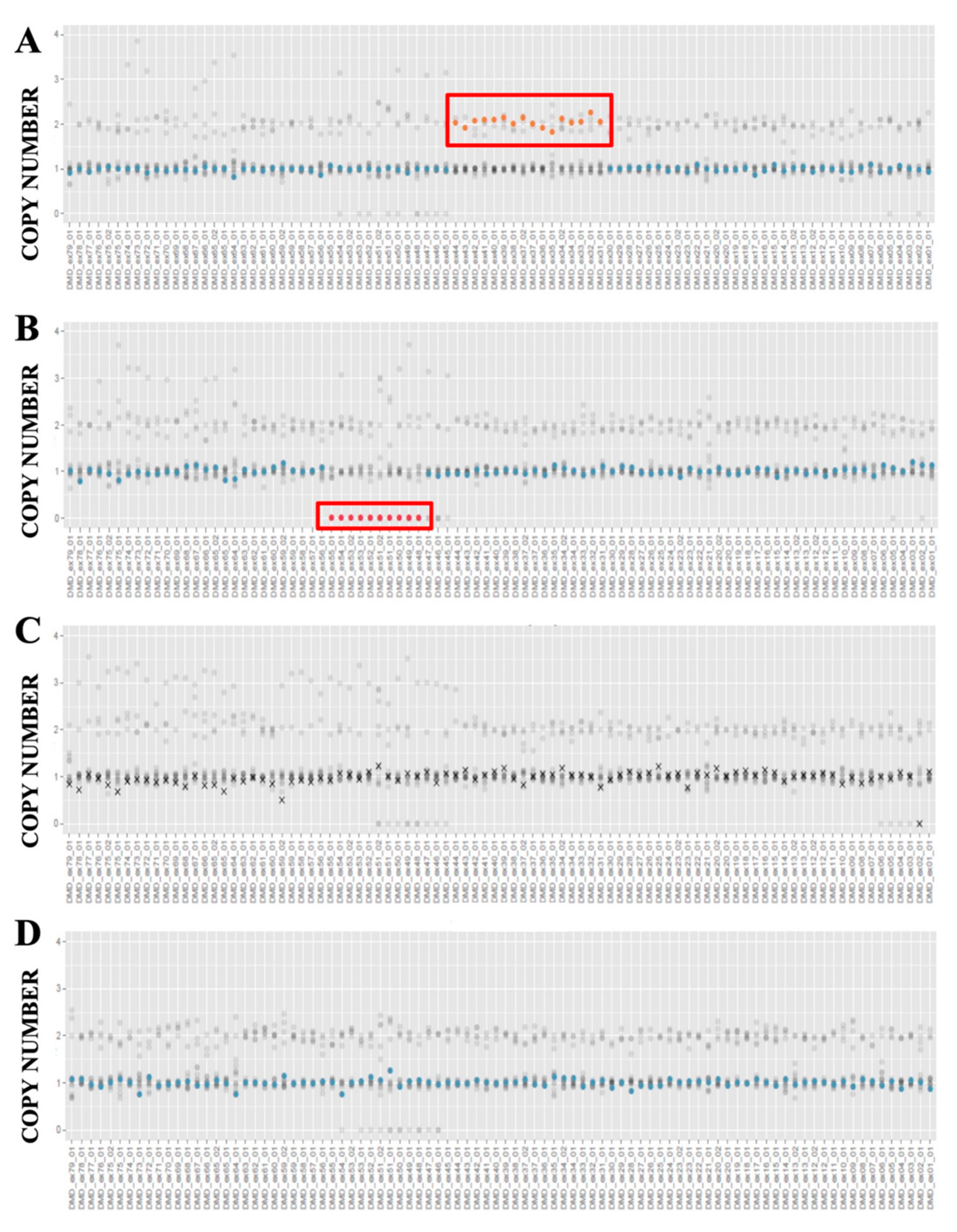
3.4. Integrative Genome Viewer (IGV) Analysis
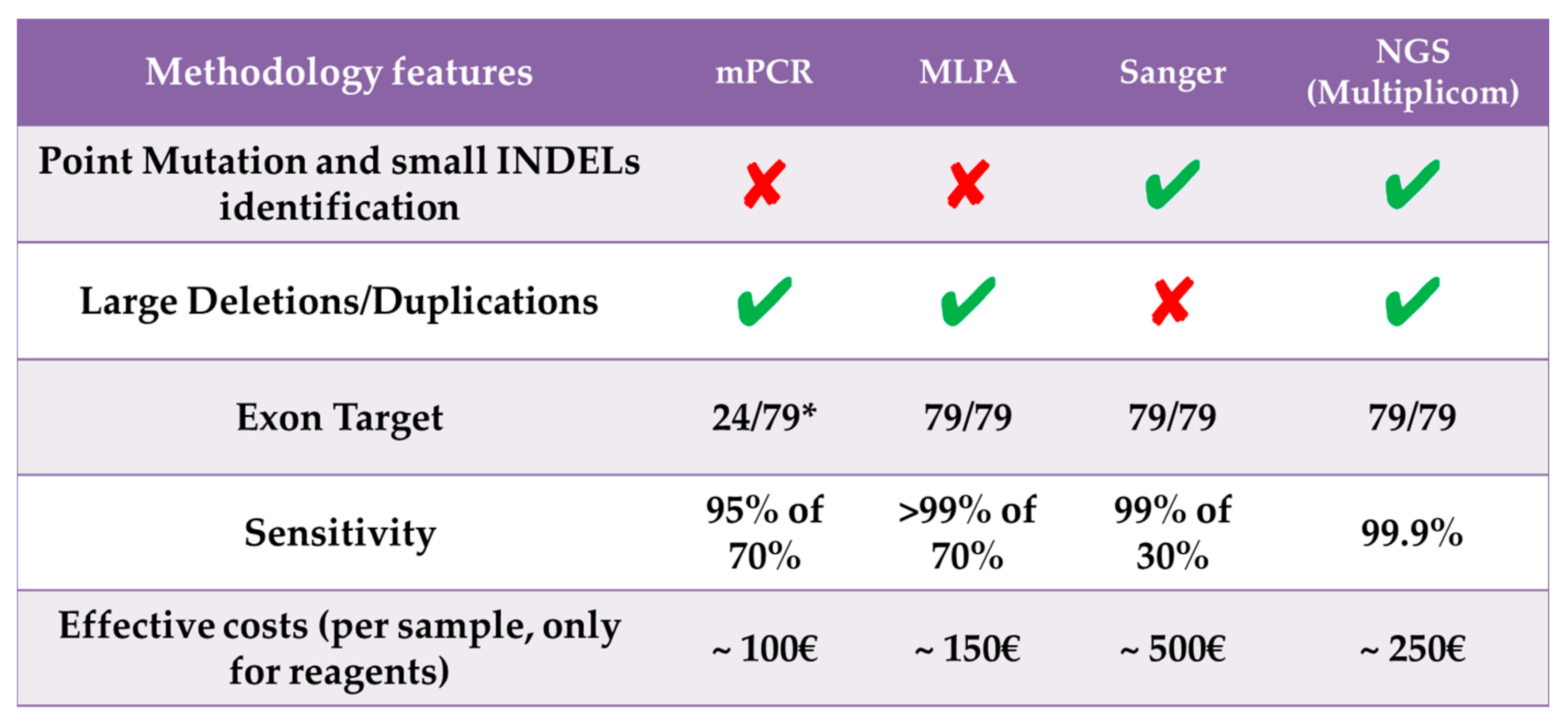
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Flanigan, K.M. Update in Duchenne and Becker muscular dystrophy. Curr. Opin. Neurol. 2019, 32, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Carsana, A. Metabolic Alterations in Cardiomyocytes of Patients with Duchenne and Becker Muscular Dystrophies. J. Clin. Med. 2019, 8, 2151. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, K.; Bougouin, W.; Sharifzadehgan, A.; Waldmann, V.; Karam, N.; Marijon, E.; Jouven, X. Sudden Cardiac Death During Sports Activities in the General Population. Card. Electrophysiol. Clin. 2017, 9, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Paolella, G.; Pisano, P.; Albano, R.; Cannaviello, L.; Mauro, C.; Esposito, G.; Vajro, P. Fatty liver disease and hypertransaminasemia hiding the association of clinically silent Duchenne muscular dystrophy and hereditary fructose intolerance. Ital. J. Pediatr. 2012, 38, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne muscular dystrophy: From diagnosis to therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wein, N.; Alfano, L.; Flanigan, K.M. Genetics and Emerging Treatments for Duchenne and Becker Muscular Dystrophy. Pediatr. Clin. N. Am. 2015, 62, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Le Rumeur, E.; Rumeur, E. Le Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn. J. Basic Med. Sci. 2015, 15, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Esposito, G.; Tremolaterra, M.R.; Marsocci, E.; Tandurella, I.C.; Fioretti, T.; Savarese, M.; Carsana, A. Precise mapping of 17 deletion breakpoints within the central hotspot deletion region (introns 50 and 51) of the DMD gene. J. Hum. Genet. 2017, 62, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Crone, M.; Mah, J.K. Current and Emerging Therapies for Duchenne Muscular Dystrophy. Curr Treat. Options Neurol. 2018, 20, 31. [Google Scholar] [CrossRef]
- Nallamilli, B.R.R.; Ankala, A.; Hegde, M. Molecular diagnosis of duchenne muscular dystrophy. Curr. Protoc. Hum. Genet. 2014, 83, 9–25. [Google Scholar] [CrossRef]
- Fratter, C.; Dalgleish, R.; Allen, S.K.; Santos, R.; Abbs, S.; Tuffery-Giraud, S.; Ferlini, A. EMQN best practice guidelines for genetic testing in dystrophinopathies. Eur. J. Hum. Genet. 2020, 28, 1141–1159. [Google Scholar] [CrossRef]
- Esposito, G.; Ruggiero, R.; Savarese, M.; Savarese, G.; Tremolaterra, M.R.; Salvatore, F.; Carsana, A. Prenatal molecular diagnosis of inherited neuromuscular diseases: Duchenne/Becker muscular dystrophy, myotonic dystrophy type 1 and spinal muscular atrophy. Clin. Chem. Lab. Med. 2013, 51, 2239–2245. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Pegoraro, E. Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2016, 35, 122–127. [Google Scholar]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genomics Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef]
- McInerney-Leo, A.M.; Duncan, E.L. Massively Parallel Sequencing for Rare Genetic Disorders: Potential and Pitfalls. Front. Endocrinol. 2021, 11, 628946. [Google Scholar] [CrossRef]
- Cock-Rada, A.M.; Ossa, C.A.; Garcia, H.I.; Gomez, L.R. A multi-gene panel study in hereditary breast and ovarian cancer in Colombia. Fam. Cancer 2018, 17, 23–30. [Google Scholar] [CrossRef]
- Nunziato, M.; Esposito, M.V.; Starnone, F.; Diroma, M.A.; Calabrese, A.; del Monaco, V.; Buono, P.; Frasci, G.; Botti, G.; D’Aiuto, M.; et al. A multi-gene panel beyond BRCA1/BRCA2 to identify new breast cancer-predisposing mutations by a picodroplet PCR followed by a next-generation sequencing strategy: A pilot study. Anal. Chim. Acta 2018, 1046, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Nerakh, G.; Ranganath, P.; Murugan, S. Next-Generation Sequencing in a Cohort of Asian Indian Patients with the Duchenne Muscular Dystrophy Phenotype: Diagnostic Yield and Mutation Spectrum. J. Pediatr. Genet. 2021, 10, 023–028. [Google Scholar] [CrossRef]
- Fioretti, T.; Auricchio, L.; Piccirillo, A.; Vitiello, G.; Ambrosio, A.; Cattaneo, F.; Ammendola, R.; Esposito, G. Multi-gene next-generation sequencing for molecular diagnosis of autosomal recessive congenital ichthyosis: A genotype-phenotype study of four Italian patients. Diagnostics 2020, 10, 995. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tao, T.; Zhao, L.; Li, G.; Yang, L. Molecular diagnosis based on comprehensive genetic testing in 800 Chinese families with non-syndromic inherited retinal dystrophies. Clin. Exp. Ophthalmol. 2021, 49, 46–59. [Google Scholar] [CrossRef]
- Yao, R.; Yu, T.; Qing, Y.; Wang, J.; Shen, Y. Evaluation of copy number variant detection from panel-based next-generation sequencing data. Mol. Genet. Genom. Med. 2019, 7, e00513. [Google Scholar] [CrossRef] [Green Version]
- Nallamilli, B.R.R.; Chaubey, A.; Valencia, C.A.; Stansberry, L.; Behlmann, A.M.; Ma, Z.; Mathur, A.; Shenoy, S.; Ganapathy, V.; Jagannathan, L.; et al. A single NGS-based assay covering the entire genomic sequence of the DMD gene facilitates diagnostic and newborn screening confirmatory testing. Hum. Mutat. 2021, 42, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Nunziato, M.; Starnone, F.; Lombardo, B.; Pensabene, M.; Condello, C.; Verdesca, F.; Carlomagno, C.; De Placido, S.; Pastore, L.; Salvatore, F.; et al. Fast detection of a BRCA2 large genomic duplication by next generation sequencing as a single procedure: A case report. Int. J. Mol. Sci. 2017, 18, 2487. [Google Scholar] [CrossRef] [Green Version]
- Carsana, A.; Frisso, G.; Intrieri, M.; Tremolaterra, M.R.; Savarese, G.; Scapagnini, G.; Esposito, G.; Santoro, L.; Salvatore, F. A 15-year molecular analysis of DMD/BMD: Genetic features in a large cohort. Front. Biosci.-Elit. 2010, 2, 547–558. [Google Scholar] [CrossRef]
- Okubo, M.; Minami, N.; Goto, K.; Goto, Y.; Noguchi, S.; Mitsuhashi, S.; Nishino, I. Genetic diagnosis of Duchenne/Becker muscular dystrophy using next-generation sequencing: Validation analysis of DMD mutations. J. Hum. Genet. 2016, 61, 483–489. [Google Scholar] [CrossRef]
- Dinh, L.T.; Tran, V.K.; Luong, L.H.; Le, P.T.; Nguyen, A.D.; Thi Nguyen, B.S.; Chi, D.V.; Tran, T.H.; Bui, T.H.; Van Ta, T.; et al. Assessment of 6 STR loci for prenatal diagnosis of Duchenne Muscular Dystrophy. Taiwan J. Obstet. Gynecol. 2019, 58, 645–649. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, D.; Liu, G.; Wang, Y.; Liu, A.; Li, L.; Luo, C.; Hu, P.; Xu, Z. Genetic analysis of 62 Chinese families with Duchenne muscular dystrophy and strategies of prenatal diagnosis in a single center. BMC Med. Genet. 2019, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Lian, Y.; Yan, Z.; Zhai, F.; Yang, M.; Zhu, X.; Wang, Y.; Nie, Y.; Guan, S.; Kuo, Y.; et al. Clinical application of an NGS-based method in the preimplantation genetic testing for Duchenne muscular dystrophy. J. Assist. Reprod. Genet. 2021, 38, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimzadeh-Vesal, R.; Teymoori, A.; Aziminezhad, M.; Hosseini, F.S. Next Generation Sequencing approach to molecular diagnosis of Duchenne muscular dystrophy; identification of a novel mutation. Gene 2018, 644, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Zhong, X.; Liu, L.; Cui, S.; Yang, Y.; Kong, L. Genetic analysis of 1051 Chinese families with Duchenne/Becker Muscular Dystrophy. BMC Med. Genet. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HGVS * Coding (cDNA) | HGVS * Protein Level | Reference SNP ID Number (rs) | Clinical Significance (ClinVar) | ACMG Classification ** | DANN Score § | MAF # |
|---|---|---|---|---|---|---|
| c.94-9dupT | rs3834997 | Benign/likely benign | Benign | NA | 0.0923 | |
| c.530+7A>T | rs72470523 | Likely Benign | Likely Benign | 0.63 | 0.00007 | |
| c.583C>T | p.Arg195 * | rs398123999 | Pathogenic | Pathogenic | 0.99 | NA |
| c.733A>G | p.Ile245Val | rs140510985 | UCV | UCV | 0.98 | 0.00004 |
| c.832A>G | p.Ile278Val | rs779964937 | NR. | UCV | 0.99 | 0.0000135 |
| c.1483-67A>T | rs1435727 | Likely Benign | Benign | 0.29 | 0.039 | |
| c.1483-72T>C | rs17309542 | NR | Benign | 0.0698 | ||
| c.1483-110G>A | rs808543 | Benign | Benign | 0.47 | 0.518 | |
| c.1635A>G | p.Arg545= | rs5927083 | Benign/likely benign | Benign | 0.54 | 0.107 |
| c.1704+51T>C | rs5927082 | Benign | Benign | 0.33 | 0.0968 | |
| c.1812C>T | p.Ala604= | rs140919039 | Benign/likely benign | Benign | 0.19 | 0.000268 |
| c.1993-37T>G | rs115571 | Benign | Benign | 0.75 | 0.724 | |
| c.2168+13T>C | rs228373 | Benign/likely benign | Benign | 0.77 | 0.313 | |
| c.2414C>G | p.Ser805 * | NR | NR | Pathogenic | 0.99 | NA |
| c.2645A>G | p.Asp882Gly | rs228406 | Benign | Benign | 0.80 | 0.728 |
| c.2827C>T | p.Arg943Cys | rs199986217 | Benign/likely benign | Benign | 0.99 | 0.000191 |
| c.3259C>T | p.Gln1087 * | rs886039536 | Pathogenic | Pathogenic | 0.99 | NA |
| c.3603+15dupA | rs5902031 | Likely benign | UCV | NA | 0.428 | |
| c.3603+15delA | rs745638361 | Benign | UCV | NA | 0.0902 | |
| c.3787-18T>C | rs72468656 | Benign | Benign | 0.76 | 0.00633 | |
| c.4120delG | p.Glu1374Argfs*8 | NR | NR | Pathogenic | NA | NA |
| c.4234-13A>G | rs41303181 | Benign/likely benign | Benign | 0.31 | 0.0607 | |
| c.4519-34T>A | rs72468639 | Benign | Benign | 0.56 | 0.0196 | |
| c.4675-53G>T | rs72468636 | Likely benign | Benign | 0.26 | 0.0115 | |
| c.5155-63T>A | rs12387861 | NR | Benign | 0.57 | 0.0346 | |
| c.5234G>A | p.Arg1745His | rs1801187 | Benign/likely benign | Benign | 0.99 | 0.528 |
| c.5326-54A>C | rs41309607 | Likely benign | Benign | 0.83 | 0.00777 | |
| c.5586+94_5586+95dupCT | rs5902025 | NR | Benign | NA | 0.767 | |
| c.5697dupA | p.Leu1900Ilefs*6 | rs794727661 | Pathogenic | Pathogenic | NA | NA |
| c.5739+15G>T | rs398123996 | NR | UCV | 0.77 | 0.0000164 | |
| c.5740-67G>T | rs6527184 | NR | Benign | 0.79 | 0.169 | |
| c.5922+11A>C | rs1394206261 | Likely benign | Likely benign | 0.78 | 0.00000561 | |
| c.6118-63_6118-62dupAT | rs3842480 | NR | Benign | NA | 0.25 | |
| c.6290+27T>A | rs3788896 | NR | Benign | 0.45 | 0.119 | |
| c.6291-115G>A | rs3747400 | Benign | Benign | 0.28 | 0.273 | |
| c.6463C>T | p.Arg2155Trp | rs1800273 | Benign/likely benign | Benign | 0.99 | 0.0261 |
| c.6614+26G>T | rs3761604 | Benign | Benign | 0.49 | 0.329 | |
| HGVS * Coding (cDNA) | HGVS * Protein Level | Reference SNP ID Number (rs) | Clinical Significance (ClinVar) | ACMGClassification ** | DANN Score § | MAF # |
| c.7096A>C | p.Gln2366Lys | rs1800275 | Benign | Benign | 0.98 | 0.234 |
| c.7310-36C>T | rs72466586 | Benign | Benign | 0.83 | 0.011 | |
| c.7542+13A>G | rs72466585 | Benign | Benign | 0.61 | 0.00384 | |
| c.7728T>C | p.Asn2576= | rs1801188 | Benign/likely benign | Benign | 0.66 | 0.17 |
| c.8027+11C>T | rs2270672 | Benign/likely benign | Benign | 0.67 | 0.332 | |
| c.8669-75C>G | rs17338583 | Benign | Benign | 0.53 | 0.0723 | |
| c.8729A>T | p.Glu2910Val | rs41305353 | Benign/likely benign | Benign | 0.99 | 0.0209 |
| c.8734A>G | p.Asn2912Asp | rs1800278 | Benign/likely benign | Benign | 0.98 | 0.0213 |
| c.8762A>G | p.His2921Arg | rs1800279 | Benign/likely benign | Benign | 0.69 | 0.0256 |
| c.8810G>A | p.Gln2937Arg | rs1800280 | Benign | Benign | 0.83 | 0.898 |
| c.9164-145A>G | rs7059188 | Benign | Benign | 0.82 | 0.071 | |
| c.9563+118C>A | NR | NR | UCV | 0.46 | NA | |
| c.9564-97C>T | rs2293667 | Benign | Benign | 0.42 | 0.843 | |
| c.9649+15T>C | rs2293668 | Benign/likely benign | Benign | 0.75 | 0.867 | |
| c.9808-63G>A | NR | NR | UCV | 0.43 | NA | |
| c.9974+22dupA | rs3833412 | NR | Benign | NA | 0.411 | |
| c.9974+22delA | rs3833412 | NR | Benign | NA | 0.114 | |
| c.10087-20C>T | rs41303187 | Benign/likely benign | Benign | 0.76 | 0.00194 | |
| c.10328+67G>A | rs2404496 | NR | Benign | 0.59 | 0.872 | |
| c.10797+42C>G | rs72466537 | NR | Benign | 0.68 | 0.00731 | |
| c.10554-36_10554-33del | rs200534475 | NR | UCV | NA | 0.746 |
| Previous Results | NGS Results | ||||||
|---|---|---|---|---|---|---|---|
| Sample ID | Gender | Phenotype | mPCR Results | MLPA Results | Sanger Point Mutation Results | NGS CNV Results | NGS Point Mutation Results * |
| 1 | M | Patient | WT | Ex13-29 dup | n.p. | Ex13-29 dup | c.8762A>G p.His2921Arg |
| 2 | M | Patient | WT | Ex57-65 dup | n.p. | Ex57-65 dup | - |
| 3 | M | Patient | WT | Ex10-11 dup | n.p. | UNDETERMINED Ex10 | - |
| 4 | F | Carrier | WT | Ex44-59;64-79 dup | n.p. | Ex44-59;64-79 dup | c.7310-36C>T (het); c.9808-63G>A (het) |
| 5 | M | Patient | WT | WT | n.p. | WT | c.583C>T (p.Arg195 *) |
| 8 | M | Patient | WT | Ex54 dup | n.p. | UNDETERMINED Ex54 | - |
| 9 | M | Patient | WT | Ex54dup | n.p. | UNDETERMINED Ex54 | - |
| 10 | M | Patient | WT | Ex2 dup | n.p. | UNDETERMINED Ex2 | - |
| 11 | M | Patient | WT | Ex55 del | n.p. | Ex55 del | - |
| 12 | F | Carrier | n.p. | n.p. | c.2414C>G (p.Ser805*) | WT | c.2414C>G (p.Ser805*); c.4519-34T>A (het); c.4675-53G>T (het); c.5155-63T>A (het) |
| 13 | M | Patient | WT | WT | c.5697dupA (p.Leu1900Ilefs*6) | WT | c.5697dupA (p.Leu1900Ilefs*6) |
| 14 | M | Patient | n.p. | n.p. | c.4120delG (p.Glu1374Argfs*8) | WT | c.4120delG (p.Glu1374Argfs*8); c.9563+118C>A |
| 15 | F | Carrier | Ex44 del | n.p. | n.p. | Ex44 del | c.5326-54A>C (het); c.8729A>T (p.Glu2910Val) (het); c.8734A>G (p.Asn2912Asp) (het); c.10797+42C>G (het) |
| 16 | M | Patient | Ex13;17;19 dup | Ex13-29 dup | n.p. | Ex13-29 dup | c.8762A>G (p.His2921Arg) |
| 17 | M | Patient | Ex48 del | Ex48 del | n.p. | Ex48 del | c.8762A>G (p.His2921Arg) |
| 18 | M | Patient | Ex41-44 dup | Ex31-44 dup | n.p. | Ex31-44 dup | c.3787-18T>C; c.8762A>G (p.His2921Arg) |
| 19 | M | Patient | Ex2 dup | Ex2 dup | n.p. | UNDETERMINED Ex2;75 | c.733A>G (p.Ile245Val) |
| 20 | M | Patient | Ex45-47;50-53 dup | Ex45-47;50-62 dup | n.p. | Ex45-47;50-62 dup | c.5326-54A>C |
| 22 | M | Patient | Ex12;13;17 dup | Ex10-17 dup | n.p. | Ex10-17 dup | c.1483-67A>T |
| 23 | F | Carrier | n.p. | n.p. | c.3259C>T (p.Gln1087*) | WT | c.3259C>T (p.Gln1087*) (het); c.832A>G (p.Ile278Val) (het) |
| 24 | M | Patient | Ex48-53 del | Ex48-55 del | n.p. | Ex48-55 del | - |
| 25 | M | Patient | Ex45-53 del | Ex45-55 del | n.p. | Ex45-55 del | - |
| 26 | M | Patient | Ex45-52 del | n.p. | n.p. | Ex45-52 del | - |
| 28 | M | Patient | Ex45-47del | n.p. | n.p. | REJECTED | c.7310-36C>T |
| 29 | F | Carrier | Ex45-48 del | Ex45-48 del | n.p. | Ex45-48 del | c.6463C>T (p.Arg2155Trp) (homo); c.7542+13A>G (het); c.8762A>G (p.His2921Arg) (het) |
| 30 | F | Carrier | n.p. | Ex26-30 del | n.p. | Ex13;26-30 del | c.1483-67A>T (het); c.4519-34T>A (het); c.5155-63T>A (het) |
| 31 | F | Carrier | Ex45-52 del | n.p. | n.p. | Ex45-52 del | - |
| 32 | F | Carrier | Ex46-48 del | n.p. | n.p. | Ex46-48 del | - |
| 34 | M | Patient | Ex17;19 dup | Ex14-20 dup | n.p. | Ex14-20 dup | - |
| 35 | F | Carrier | Ex12-19 del | Ex10-29 del | n.p. | Ex10-29 del | - |
| 38 | M | Patient | n.p. | Ex48 del | n.p. | Ex48 del | c.2827C>T (p.Arg943Cys); c.3787-18T>C; c.8762A>G (p.His2921Arg) |
| 40 | M | Patient | Ex45-52 del | Ex45-55 del | n.p. | Ex45-55 del | - |
| 41 | M | Patient | Ex45-47 del | Ex45-47 del | n.p. | Ex45-47 del | c.530+7A>T; c.1812C>T (p.Ala604=) |
| 42 | M | Patient | Ex48-51 del | Ex48-51 del | n.p. | Ex48-51 del | c.10087-20C>T |
| 43 | M | Patient | Ex2 del | Ex2 del | n.p. | REJECTED | - |
| 44 | M | Patient | Ex2-6 del | Ex2-7 del | n.p. | REJECTED | - |
| 45 | M | Patient | Ex46-51 del | Ex46-51 del | n.p. | Ex46-51 del | c.4519-34T>A; c.4675-53G>T; c.5155-63T>A; c.10087-20C>T |
| 46 | M | Patient | n.p. | n.p. | n.p. | WT | c.4519-34T>A; c.4675-53G>T; c.5155-63T>A |
| 47 | M | Patient | n.p. | n.p. | n.p. | WT | - |
| 48 | M | Patient | n.p. | n.p. | n.p. | REJECTED | c.5326-54A>C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Palma, F.D.E.; Nunziato, M.; D’Argenio, V.; Savarese, M.; Esposito, G.; Salvatore, F. Comprehensive Molecular Analysis of DMD Gene Increases the Diagnostic Value of Dystrophinopathies: A Pilot Study in a Southern Italy Cohort of Patients. Diagnostics 2021, 11, 1910. https://doi.org/10.3390/diagnostics11101910
De Palma FDE, Nunziato M, D’Argenio V, Savarese M, Esposito G, Salvatore F. Comprehensive Molecular Analysis of DMD Gene Increases the Diagnostic Value of Dystrophinopathies: A Pilot Study in a Southern Italy Cohort of Patients. Diagnostics. 2021; 11(10):1910. https://doi.org/10.3390/diagnostics11101910
Chicago/Turabian StyleDe Palma, Fatima Domenica Elisa, Marcella Nunziato, Valeria D’Argenio, Maria Savarese, Gabriella Esposito, and Francesco Salvatore. 2021. "Comprehensive Molecular Analysis of DMD Gene Increases the Diagnostic Value of Dystrophinopathies: A Pilot Study in a Southern Italy Cohort of Patients" Diagnostics 11, no. 10: 1910. https://doi.org/10.3390/diagnostics11101910
APA StyleDe Palma, F. D. E., Nunziato, M., D’Argenio, V., Savarese, M., Esposito, G., & Salvatore, F. (2021). Comprehensive Molecular Analysis of DMD Gene Increases the Diagnostic Value of Dystrophinopathies: A Pilot Study in a Southern Italy Cohort of Patients. Diagnostics, 11(10), 1910. https://doi.org/10.3390/diagnostics11101910