
Next Generation Sequencing of Cerebrospinal Fluid B Cell Repertoires in Multiple Sclerosis and Other Neuro-Inflammatory Diseases—A Comprehensive Review

 ,
,
Abstract
:1. Introduction
1.1. Neuro-Inflammatory Diseases—Cerebrospinal Fluid (CSF) Findings, Including Routine Diagnostics, Autoantibodies and B Cells
1.2. B Cell Repertoires—The CDR3 Region as a Molecular Fingerprint of B Cell Maturation
1.3. Next-Generation Sequencing of B Cell Repertoires—Methods and Data Analysis
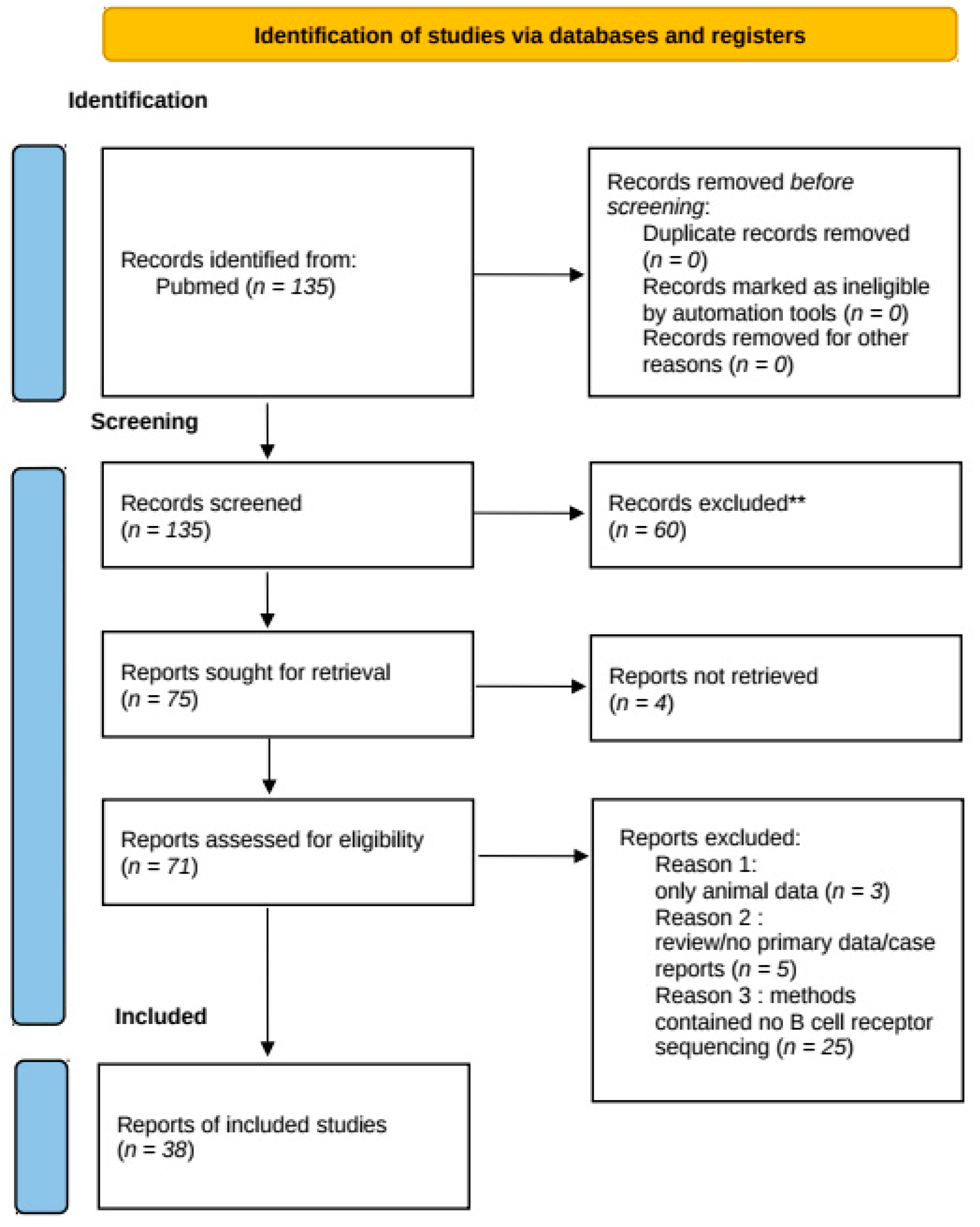
2. Methods of Systematic Review
3. Results
3.1. CSF B Cell Repertoires in Multiple Sclerosis
3.1.1. Enrichment of VH4 Family Usage in CSF B Cell Repertoires
3.1.2. Clonal Expansion of B Cells within the CSF Compartment
3.1.3. Spatial Tracking of B Cell Clones over the BBB—Bidirectional Exchange of B Cells
3.1.4. Temporal Tracking of B Cell Clones within the CSF Compartment: Treatment Specific Effects on B Cell Clones
3.1.5. Overlap between Ig Transcriptome and Proteome: Evidence for CSF B Cells as the Origin of Intrathecal Ig
3.2. CSF B Cell Repertoires in Autoantibody-Mediated Encephalitis
3.2.1. Neuromyelitis Optica Spectrum Disorders
3.2.2. LGI1 and NMDAR Autoantibody-Mediated Encephalitis
4. Discussion
- Regarding the analysis of basic B cell repertoire characteristics, a preferential usage of the VH4 germline family within CSF B cell repertoires has been consistently shown in several studies. These data suggest that a chronic B cell stimulation with a common mechanism or antigen(s) might occur with epitopes that are preferentially recognized by the hypervariable loop structure of VH4 segments.
- These results go along with the consistent finding from both single cell and NGS CSF B cell repertoire analyses, indicating that a substantial proportion of CSF B cell clones undergo CSF compartment-specific maturation steps, including clonal expansion.
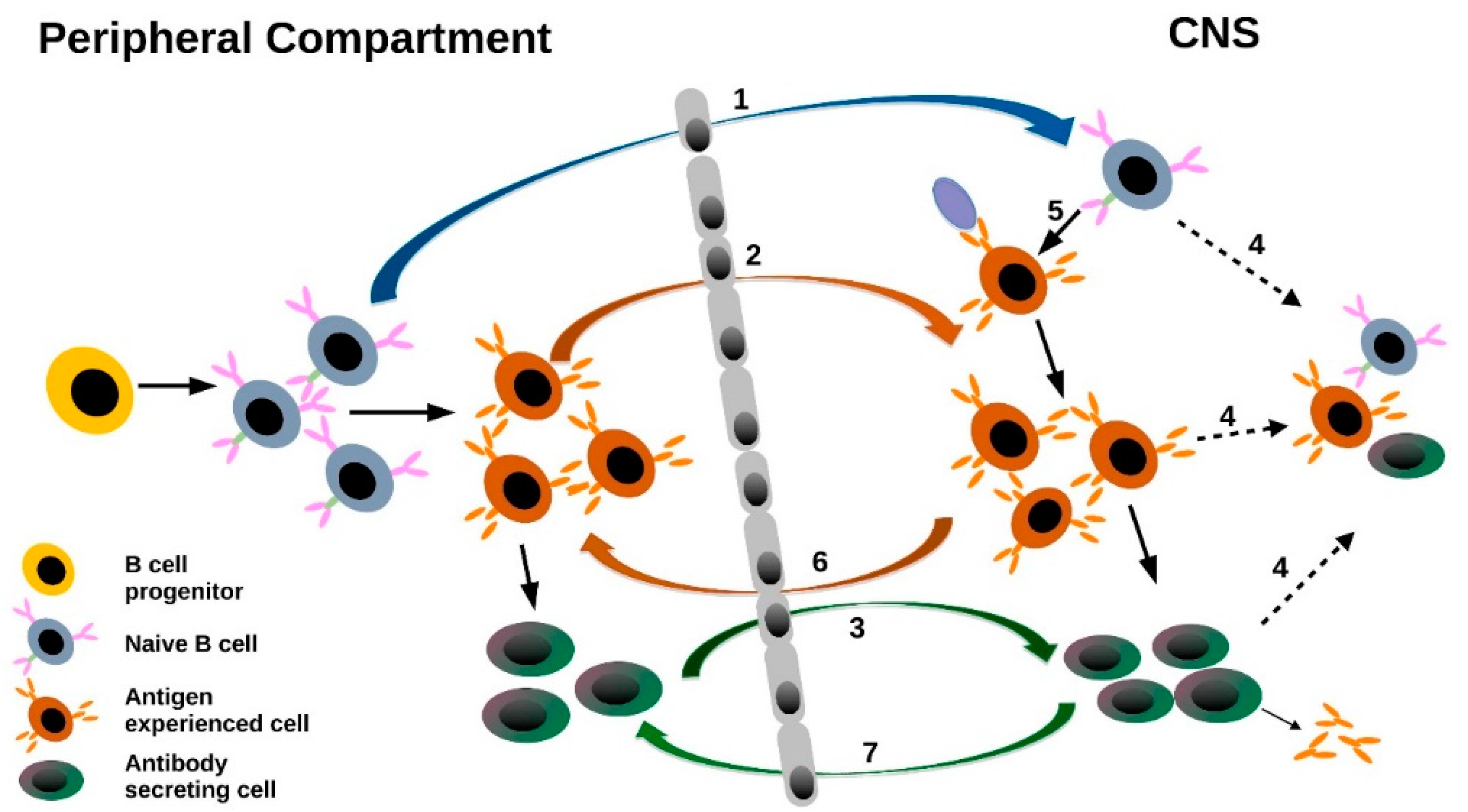
- The most striking results derived from NGS studies on CSF and PB B cell repertoires relate to B cell trafficking across the BBB. An intense exchange of B cells across the BBB is observed in MS and other autoimmune disorders, pointing towards an involvement of additional compartments outside of the CNS. Studies consistently show that B cells not only migrate into the CSF but also seem to leave the CSF compartment to undergo further maturation in the periphery. Although a direct proof for B cell trafficking patterns would require in vivo B cell tracking, several studies show that B cells in various tissues, such as cervical lymph nodes [52], connect with CNS B cells, with maturation steps occurring on both sides of the BBB. A schematic summary of B cell trafficking is displayed in Figure 2.
- Combined analyses of CSF transcriptome and proteome B cell repertoires further reveal that intrathecal Ig is indeed produced by CSF B cells/B cell clones.
5. Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sabatino, J.J.; Pröbstel, A.-K.; Zamvil, S.S. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat. Rev. Neurosci. 2019, 20, 728–745. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lepennetier, G.; Hracsko, Z.; Unger, M.; Van Griensven, M.; Grummel, V.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Kowarik, M.C. Cytokine and immune cell profiling in the cerebrospinal fluid of patients with neuro-inflammatory diseases. J. Neuroinflamm. 2019, 16, 1–11. [Google Scholar] [CrossRef]
- Kowarik, M.C.; Grummel, V.; Wemlinger, S.; Buck, D.; Weber, M.S.; Berthele, A.; Hemmer, B. Immune cell subtyping in the cerebrospinal fluid of patients with neurological diseases. J. Neurol. 2014, 261, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Astling, D.; Gasperi, C.; Wemlinger, S.; Schumann, H.; Dzieciatkowska, M.; Ritchie, A.M.; Hemmer, B.; Owens, G.P.; Bennett, J.L. CNS Aquaporin-4-Specific B Cells Connect with Multiple B-Cell Compartments in Neuromyelitis Optica Spectrum Disorder. Ann. Clin. Transl. Neurol. 2017, 4, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef]
- Obermeier, B.; Mentele, R.; Malotka, J.; Kellermann, J.; Kümpfel, T.; Wekerle, H.; Lottspeich, F.; Hohlfeld, R.; Dornmair, K. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008, 14, 688–693. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Cepok, S.; Rosche, B.; Grummel, V.; Vogel, F.; Zhou, D.; Sayn, J.; Sommer, N.; Hartung, H.-P.; Hemmer, B. Short-lived plasma blasts are the main b cell effector subset during the course of multiple sclerosis. Brain 2005, 128, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, V.A.; Pittock, S.J.; Lucchinetti, C.F.; Weinshenker, B.G. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006, 66, 1485–1489. [Google Scholar] [CrossRef] [Green Version]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Bennett, J.L.; O’Connor, K.C.; Bar-Or, A.; Zamvil, S.S.; Hemmer, B.; Tedder, T.F.; von Büdingen, H.-C.; Stuve, O.; Yeaman, M.R.; Smith, T.J.; et al. B lymphocytes in neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Kowarik, M.C.; Dzieciatkowska, M.; Wemlinger, S.; Ritchie, A.M.; Hemmer, B.; Owens, G.P.; Bennett, J.L. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J. Neuroinflamm. 2015, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Paul, F.; Weinshenker, B.G.; Levy, M.; Kim, H.J.; Wildemann, B. Neuromyelitis optica. Nat. Rev. Dis. Primers 2020, 6, 85. [Google Scholar] [CrossRef]
- Bar-Or, A.; Calabresi, P.A.J.; Arnold, D.; Arnlod, D.; Markowitz, C.; Shafer, S.; Kasper, L.H.; Waubant, E.; Gazda, S.; Fox, R.J.; et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann. Neurol. 2008, 63, 395–400. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Lisby, S.; Grove, R.; Derosier, F.; Shackelford, S.; Havrdova, E.; Drulovic, J.; Filippi, M. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: A phase 2 study. Neurology 2014, 82, 573–581. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Jacob, A.; Weinshenker, B.G.; Violich, I.; McLinskey, N.; Krupp, L.; Fox, R.J.; Wingerchuk, D.M.; Boggild, M.; Constantinescu, C.S.; Miller, A.; et al. Treatment of neuromyelitis optica with rituximab: Retrospective analysis of 25 patients. Arch. Neurol. 2008, 65, 1443–1448. [Google Scholar] [CrossRef]
- Kim, S.-H.; Huh, S.-Y.; Lee, S.J.; Joung, A.; Kim, H.J. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013, 70, 1110–1117. [Google Scholar] [CrossRef] [Green Version]
- Prüss, H. Autoantibodies in neurological disease. Nat. Rev. Immunol. 2021, 1–16. [Google Scholar] [CrossRef]
- Lehmann-Horn, K.; Irani, S.R.; Wang, S.; Palanichamy, A.; Jahn, S.; Greenfield, A.L.; Dandekar, R.; Lepennetier, G.; Michael, S.; Gelfand, J.M.; et al. Intrathecal B-cell activation in LGI1 antibody encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Ramberger, M.; O’Connor, K.C.; Bashford-Rogers, R.J.M.; Irani, S.R. The B cell immunobiology that underlies CNS autoantibody-mediated diseases. Nat. Rev. Neurol. 2020, 16, 481–492. [Google Scholar] [CrossRef]
- Gresa-Arribas, N.; Titulaer, M.J.; Torrents, A.; Aguilar, E.; McCracken, L.; Leypoldt, F.; Gleichman, A.J.; Balice-Gordon, R.; Rosenfeld, M.R.; Lynch, D.; et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: A retrospective study. Lancet Neurol. 2014, 13, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Malter, M.P.; Elger, C.E.; Surges, R. Diagnostic value of CSF findings in antibody-associated limbic and anti-NMDAR-encephalitis. Seizure 2013, 22, 136–140. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Küppers, R. Human memory B cells. Leukemia 2016, 30, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Astling, D.; Lepennetier, G.; Ritchie, A.; Hemmer, B.; Owens, G.P.; Bennett, J.L. Differential effects of fingolimod and natalizumab on B cell repertoires in multiple sclerosis patients. Neurotherapeutics 2020, 18, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, A.L.; Dandekar, R.; Ramesh, A.; Eggers, E.L.; Wu, H.; Laurent, S.; Harkin, W.; Pierson, N.S.; Weber, M.S.; Henry, R.G.; et al. Longitudinally persistent cerebrospinal fluid B cells can resist treatment in multiple sclerosis. JCI Insight 2019, 4, e126599. [Google Scholar] [CrossRef] [Green Version]
- Benichou, J.; Ben-Hamo, R.; Louzoun, Y.; Efroni, S. Rep-seq: Uncovering the immunological repertoire through next-generation sequencing. Immunology 2012, 135, 183–191. [Google Scholar] [CrossRef]
- Boyd, S.D.; Joshi, S.A. High-throughput DNA sequencing analysis of antibody repertoires. Microbiol. Spectr. 2014, 2, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Mamedov, I.Z.; Britanova, O.V.; Zvyagin, I.V.; Turchaninova, M.A.; Bolotin, D.A.; Putintseva, E.V.; Lebedev, Y.B.; Chudakov, D.M. Preparing unbiased T-cell receptor and antibody CDNA libraries for the deep next generation sequencing profiling. Front. Immunol. 2013, 4, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barennes, P.; Quiniou, V.; Shugay, M.; Egorov, E.S.; Davydov, A.N.; Chudakov, D.M.; Uddin, I.; Ismail, M.; Oakes, T.; Chain, B.; et al. Benchmarking of T cell receptor repertoire profiling methods reveals large systematic biases. Nat. Biotechnol. 2021, 39, 236–245. [Google Scholar] [CrossRef]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef]
- Smakaj, E.; Babrak, L.; Ohlin, M.; Shugay, M.; Briney, B.; Tosoni, D.; Galli, C.; Grobelsek, V.; D’Angelo, I.; Olson, B.; et al. Benchmarking immunoinformatic tools for the analysis of antibody repertoire sequences. Bioinformatics 2020, 36, 1731–1739. [Google Scholar] [CrossRef] [Green Version]
- Shlemov, A.; Bankevich, S.; Bzikadze, A.; Turchaninova, M.A.; Safonova, Y.; Pevzner, P.A. Reconstructing antibody repertoires from error-prone immunosequencing reads. J. Immunol. 2017, 199, 3369–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Santibanez-Jacome, L.; Erendira Avendano-Vazquez, S.; Fabian Flores-Jasso, C. The pipeline repertoire for Ig-seq analysis. Front. Immunol. 2019, 10, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Ma, N.; Madden, T.L.; Ostell, J.M. IgBLAST: An immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013, 41, W34–W40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Lefranc, M.-P.; Miles, J.J.; Alamyar, E.; Giudicelli, V.; Duroux, P.; Freeman, J.D.; Corbin, V.D.A.; Scheerlinck, J.-P.; Frohman, M.A.; et al. IMGT/HighV QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat. Commun. 2013, 4, 2333. [Google Scholar] [CrossRef]
- Lefranc, M.-P.; Giudicelli, V.; Duroux, P.; Jabado-Michaloud, J.; Folch, G.; Aouinti, S.; Carillon, E.; Duvergey, H.; Houles, A.; Paysan-Lafosse, T.; et al. IMGT®, the International ImMunoGeneTics Information System® 25 Years On. Nucleic Acids Res. 2015, 43, D413–D422. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.T.; Vander Heiden, J.A.; Uduman, M.; Gadala-Maria, D.; Yaari, G.; Kleinstein, S.H. Change-O: A toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 2015, 31, 3356–3358. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbecker, L.; Nienen, M.; Hecht, J.; Neumann, A.U.; Babel, N.; Reinert, K.; Robinson, P.N. IMSEQ—A fast and error aware approach to immunogenetic sequence analysis. Bioinformatics 2015, 31, 2963–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortina-Ceballos, B.; Godoy-Lozano, E.E.; Sámano-Sánchez, H.; Aguilar-Salgado, A.; Velasco-Herrera, M.D.C.; Vargas-Chávez, C.; Velázquez-Ramírez, D.; Romero, G.; Moreno, J.; Téllez-Sosa, J.; et al. Reconstructing and mining the B cell repertoire with immunediversity. MAbs 2015, 7, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.O. Diversity and evenness: A unifying notation and its consequences. Ecology 1973, 54, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Owens, G.P.; Kannus, H.; Burgoon, M.P.; Smith-Jensen, T.; Devlin, M.E.; Gilden, D.H. Restricted use of VH4 Germline segments in an acute multiple sclerosis brain. Ann. Neurol. 1998, 43, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.N.H.; Yaari, G.; Vander Heiden, J.A.; Church, G.; Donahue, W.F.; Hintzen, R.Q.; Huttner, A.J.; Laman, J.D.; Nagra, R.M.; Nylander, A.; et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014, 6, 248ra107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.P.; Ritchie, A.M.; Gilden, D.H.; Burgoon, M.P.; Becker, D.; Bennett, J.L. Measles virus–specific plasma cells are prominent in subacute sclerosing panencephalitis CSF. Neurology 2007, 68, 1815–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Duquette, P.; Zhang, Y.; Talbot, P.; Poole, R.; Antel, J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J. Clin. Investig. 1998, 102, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- von Büdingen, H.-C.; Kuo, T.C.; Sirota, M.; van Belle, C.J.; Apeltsin, L.; Glanville, J.; Cree, B.A.; Gourraud, P.-A.; Schwartzburg, A.; Huerta, G.; et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J. Clin. Investig. 2012, 122, 4533–4543. [Google Scholar] [CrossRef]
- Beltrán, E.; Obermeier, B.; Moser, M.; Coret, F.; Simó-Castelló, M.; Boscá, I.; Pérez-Miralles, F.; Villar, L.M.; Senel, M.; Tumani, H.; et al. Intrathecal somatic hypermutation of IgM in multiple sclerosis and neuroinflammation. Brain 2014, 137, 2703–2714. [Google Scholar] [CrossRef] [Green Version]
- Palanichamy, A.; Apeltsin, L.; Kuo, T.C.; Sirota, M.; Wang, S.; Pitts, S.J.; Sundar, P.D.; Telman, D.; Zhao, L.Z.; Derstine, M.; et al. Immunoglobulin class-switched B cells provide an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 2014, 6, 248ra106. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.L.; Haubold, K.; Ritchie, A.M.; Edwards, S.J.; Burgoon, M.; Shearer, A.J.; Gilden, D.H.; Owens, G.P. CSF IgG heavy-chain bias in patients at the time of a clinically isolated syndrome. J. Neuroimmunol. 2008, 199, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Baranzini, S.E.; Jeong, M.C.; Butunoi, C.; Murray, R.S.; Bernard, C.C.A.; Oksenberg, J.R. B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J. Immunol. 1999, 163, 5133–5144. [Google Scholar]
- Smith–Jensen, T.; Burgoon, M.P.; Anthony, J.; Kraus, H.; Gilden, D.H.; Owens, G.P. Comparison of Immunoglobulin G Heavy-chain sequences in MS and SSPE brains reveals an antigen-driven response. Neurology 2000, 54, 1227–1232. [Google Scholar] [CrossRef]
- Monson, N.L.; Brezinschek, H.-P.; Brezinschek, R.I.; Mobley, A.; Vaughan, G.K.; Frohman, E.M.; Racke, M.K.; Lipsky, P.E. Receptor revision and atypical mutational characteristics in clonally expanded B cells from the cerebrospinal fluid of recently diagnosed multiple sclerosis patients. J. Neuroimmunol. 2005, 158, 170–181. [Google Scholar] [CrossRef]
- Owens, G.P.; Burgoon, M.P.; Anthony, J.; Kleinschmidt-DeMasters, B.K.; Gilden, D.H. The immunoglobulin g heavy chain repertoire in multiple sclerosis plaques is distinct from the heavy chain repertoire in peripheral blood lymphocytes. Clin. Immunol. 2001, 98, 258–263. [Google Scholar] [CrossRef]
- Johansen, J.N.; Vartdal, F.; Desmarais, C.; Tutturen, A.E.V.; de Souza, G.A.; Lossius, A.; Holmøy, T. Intrathecal BCR transcriptome in multiple sclerosis versus other neuroinflammation: Equally diverse and compartmentalized, but more mutated, biased and overlapping with the proteome. Clin. Immunol. 2015, 160, 211–225. [Google Scholar] [CrossRef]
- Vander Heiden, J.A.; Yaari, G.; Uduman, M.; Stern, J.N.H.; O’Connor, K.C.; Hafler, D.A.; Vigneault, F.; Kleinstein, S.H. PRESTO: A toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 2014, 30, 1930–1932. [Google Scholar] [CrossRef]
- Nouri, N.; Kleinstein, S.H. Optimized threshold inference for partitioning of clones from high-throughput B cell repertoire sequencing data. Front. Immunol. 2018, 9, 1687. [Google Scholar] [CrossRef] [Green Version]
- Yaari, G.; Uduman, M.; Kleinstein, S.H. Quantifying selection in high-throughput immunoglobulin sequencing data sets. Nucleic Acids Res. 2012, 40, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barak, M.; Zuckerman, N.S.; Edelman, H.; Unger, R.; Mehr, R. IgTree©: Creating immunoglobulin variable region gene lineage trees. J. Immunol. Methods 2008, 338, 67–74. [Google Scholar] [CrossRef]
- Glanville, J.; Zhai, W.; Berka, J.; Telman, D.; Huerta, G.; Mehta, G.R.; Ni, I.; Mei, L.; Sundar, P.D.; Day, G.M.R.; et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA 2009, 106, 20216–20221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Dono, M.; Gazzola, P.; Roncella, S.; Valetto, A.; Chiorazzi, N.; Mancardi, G.L.; Ferrarini, M. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. J. Immunol. 2000, 164, 2782–2789. [Google Scholar] [CrossRef]
- Owens, G.P.; Ritchie, A.M.; Burgoon, M.P.; Williamson, R.A.; Corboy, J.R.; Gilden, D.H. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J. Immunol. 2003, 171, 2725–2733. [Google Scholar] [CrossRef] [Green Version]
- Harp, C.; Lee, J.; Lambracht-Washington, D.; Cameron, E.; Olsen, G.; Frohman, E.; Racke, M.; Monson, N. Cerebrospinal fluid B cells from multiple sclerosis patients are subject to normal germinal center selection. J. Neuroimmunol. 2007, 183, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, E.M.; Spencer, S.; Lazarini, J.; Harp, C.T.; Ward, E.S.; Burgoon, M.; Owens, G.P.; Racke, M.K.; Bennett, J.L.; Frohman, E.M.; et al. Potential of a unique antibody gene signature to predict conversion to clinically definite multiple sclerosis. Adv. Neuroimmunol. 2009, 213, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Da, R.-R.; Hilgenberg, L.G.; Tourtellotte, W.W.; Sobel, R.A.; Smith, M.A.; Olek, M.; Nagra, R.; Sudhir, G.; van den Noort, S.; et al. Clonal expansion of IgA-positive plasma cells and axon-reactive antibodies in MS lesions. J. Neuroimmunol. 2005, 167, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’Connor, K.C.; Hafler, D.A. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 2011, 134, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Lovato, L.; Mentele, R.; Brück, W.; Forne, I.; Imhof, A.; Lottspeich, F.; Turk, K.W.; Willis, S.N.; Wekerle, H.; et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J. Neuroimmunol. 2011, 233, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankoti, J.; Apeltsin, L.; Hauser, S.L.; Allen, S.; Albertolle, M.E.; Witkowska, H.E.; von Büdingen, H.-C. In multiple sclerosis, oligoclonal bands connect to peripheral b-cell responses. Ann. Neurol. 2014, 75, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Eggers, E.L.; Michel, B.A.; Wu, H.; Wang, S.; Bevan, C.J.; Abounasr, A.; Pierson, N.S.; Bischof, A.; Kazer, M.; Leitner, E.; et al. Clonal relationships of CSF B cells in treatment-naive multiple sclerosis patients. JCI Insight 2017, 2, e92724. [Google Scholar] [CrossRef]
- Tomescu-Baciu, A.; Johansen, J.N.; Holmøy, T.; Greiff, V.; Stensland, M.; de Souza, G.A.; Vartdal, F.; Lossius, A. Persistence of intrathecal oligoclonal b cells and IgG in multiple sclerosis. J. Neuroimmunol. 2019, 333, 576966. [Google Scholar] [CrossRef]
- Haubold, K.; Owens, G.P.; Kaur, P.; Ritchie, A.M.; Gilden, D.H.; Bennett, J.L. B-Lymphocyte and plasma cell clonal expansion in monosymptomatic optic neuritis cerebrospinal fluid. Ann. Neurol. 2004, 56, 97–107. [Google Scholar] [CrossRef]
- Gasperi, C.; Salmen, A.; Antony, G.; Bayas, A.; Heesen, C.; Kümpfel, T.; Linker, R.A.; Paul, F.; Stangel, M.; Tackenberg, B.; et al. Association of intrathecal immunoglobulin g synthesis with disability worsening in multiple sclerosis. JAMA Neurol. 2019, 76, 841–849. [Google Scholar] [CrossRef]
- Farina, G.; Magliozzi, R.; Pitteri, M.; Reynolds, R.; Rossi, S.; Gajofatto, A.; Benedetti, M.D.; Facchiano, F.; Monaco, S.; Calabrese, M. Increased cortical lesion load and intrathecal inflammation is associated with oligoclonal bands in multiple sclerosis patients: A combined csf and mri study. J. Neuroinflamm. 2017, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- von Büdingen, H.-C.; Gulati, M.; Kuenzle, S.; Fischer, K.; Rupprecht, T.A.; Goebels, N. Clonally expanded plasma cells in the cerebrospinal fluid of patients with central nervous system autoimmune demyelination produce “oligoclonal bands”. J. Neuroimmunol. 2010, 218, 134–139. [Google Scholar] [CrossRef]
- Brändle, S.M.; Obermeier, B.; Senel, M.; Bruder, J.; Mentele, R.; Khademi, M.; Olsson, T.; Tumani, H.; Kristoferitsch, W.; Lottspeich, F.; et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 7864–7869. [Google Scholar] [CrossRef] [Green Version]
- Soltys, J.; Liu, Y.; Ritchie, A.; Wemlinger, S.; Schaller, K.; Schumann, H.; Owens, G.P.; Bennett, J.L. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J. Clin. Investig. 2019, 129, 2000–2013. [Google Scholar] [CrossRef] [Green Version]
- Chihara, N.; Aranami, T.; Oki, S.; Matsuoka, T.; Nakamura, M.; Kishida, H.; Yokoyama, K.; Kuroiwa, Y.; Hattori, N.; Okamoto, T.; et al. Plasmablasts as migratory IgG-producing cells in the pathogenesis of neuromyelitis optica. PLoS ONE 2013, 8, e83036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornau, H.-C.; Kreye, J.; Stumpf, A.; Fukata, Y.; Parthier, D.; Sammons, R.P.; Imbrosci, B.; Kurpjuweit, S.; Kowski, A.B.; Fukata, M.; et al. Human cerebrospinal fluid monoclonal lGI1 autoantibodies increase neuronal excitability. Ann. Neurol. 2020, 87, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreye, J.; Wenke, N.K.; Chayka, M.; Leubner, J.; Murugan, R.; Maier, N.; Jurek, B.; Ly, L.-T.; Brandl, D.; Rost, B.R.; et al. Human cerebrospinal fluid monoclonal n-methyl-d-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 2016, 139, 2641–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Fan, S.; Sun, Y.; Zhang, Z.; Ren, H.; Li, W.; Cui, L.; Peng, B.; Ren, X.; Zhang, W.; et al. Study of B cell repertoire in patients with anti-n-methyl-d-aspartate receptor encephalitis. Front. Immunol. 2020, 11, 1539. [Google Scholar] [CrossRef] [PubMed]
- Bashford-Rogers, R.J.M.; Bergamaschi, L.; McKinney, E.F.; Pombal, D.C.; Mescia, F.; Lee, J.C.; Thomas, D.C.; Flint, S.M.; Kellam, P.; Jayne, D.R.W.; et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 2019, 574, 122–126. [Google Scholar] [CrossRef]
- Zou, A.; Ramanathan, S.; Dale, R.C.; Brilot, F. Single-cell approaches to investigate b cells and antibodies in autoimmune neurological disorders. Cell. Mol. Immunol. 2021, 18, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Schafflick, D.; Xu, C.A.; Hartlehnert, M.; Cole, M.; Schulte-Mecklenbeck, A.; Lautwein, T.; Wolbert, J.; Heming, M.; Meuth, S.G.; Kuhlmann, T.; et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat. Commun. 2020, 11, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, A.; Schubert, R.D.; Greenfield, A.L.; Dandekar, R.; Loudermilk, R.; Sabatino, J.J.; Koelzer, M.T.; Tran, E.B.; Koshal, K.; Kim, K.; et al. A pathogenic and clonally expanded b cell transcriptome in active multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 22932–22943. [Google Scholar] [CrossRef]
- Song, E.; Bartley, C.M.; Chow, R.D.; Ngo, T.T.; Jiang, R.; Zamecnik, C.R.; Dandekar, R.; Loudermilk, R.P.; Dai, Y.; Liu, F.; et al. Divergent and self-reactive immune responses in the CNS of COVID-19 patients with neurological symptoms. Cell Rep. Med. 2021, 2, 100288. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Al-Eryani, G.; Carswell, S.; Ferguson, J.M.; Blackburn, J.; Barton, K.; Roden, D.; Luciani, F.; Giang Phan, T.; Junankar, S.; et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat. Commun. 2019, 10, 3120. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Study | Diagnosis/Number of Patients | Compartment of B Cell Analysis | Methods | VH Family Bias (within CSF) Towards: | CSF Clonal Expansion (in MS Patients) | Clones between CNS/CSF and Periphery |
|---|---|---|---|---|---|---|
| (Qin et al., 1998) | MS/n = 12 OND/n = 15 | CSF | tRT-PCR IgVH-PCR | VH4 |
• Dominant clone(s) (10/12 MS patients) • Numerous SHM | N/A |
| (Colombo et al., 2000) | MS/n = 10 OND/n = 10 | CSF/blood | tRT-PCR IgVH-PCR | VH3 VH4 | • Oligoclonal B cell accumulations (10/10 MS patients) | CSF B cells hardly represented in PB, compartmentalized clonal expansion within the CSF |
| (Owens et al., 2003) | MS/n = 4 Viral meningitis/n = 2 | CSF | ScRT-PCR, IgH/L-PCR | N/A | • B cell clonal IgG expansion (3/4 MS patients) | N/A |
| (Owens et al., 2007) | MS/n = 15 OND/n = 2 | CSF/blood | ScRT-PCR IgH/L-PCR | VH4-39 VH4-31 VH4-59 | • 64% of CSF CD138 Cells in clonal populations | N/A |
| (Bennett et al., 2008) | CIS/n = 10 | CSF/blood | ScRT-PCR IgH/L-PCR | VH4 VH2 (70%) | • Expanded B and plasma cell clonal populations | N/A |
| (H.-Christian von Büdingen et al., 2012) | MS/n = 6 OND/n = 7 | CSF/blood | NGS | IGHV4-39 IGHV4-59 IGHV4-61 | • CSF-restricted B cell activation in MS patients | • Bidirectional exchange across the BBB in a restricted pool of clonally related B cells • Clusters undergo active diversification primarily in the CNS, in the periphery or in both compartments in parallel |
| (Palanichamy et al., 2014) | MS/n = 8 | CSF/blood | NGS | IGHV4-39 IGHV4-59 VH4-61 | • B cells belonging to bicompartmental clusters may have been exposed to antigen-stimulation in the CSF or PB | • SM: most frequent immune axis between PB and CSF • Class-switched DN B cells: also clonally related to CSF Ig repertoires • PB plasma cells: few bicompartmental clusters |
| (Beltrán et al., 2014) | MS/n = 12 OND/n = 7 | CSF/blood | High-throughput pyrosequencing | VH4-34, 4-39; VH4-59; VH4-4, 4-61; VH4-31 | • Extensive SHM in CSF IgM antibodies • AICDA expressed in CSF IgM-producing B cells | Clonal tracking/lineage: • Ancestors of CSF B cell clones reside in PB • Maturation continues in CSF • No isotype switching from IgM to IgG in CSF |
| (Stern et al., 2014) | MS/n = 5 | CNS tissue, CLN | SSC/NGS | IGHV4 (CNS vs. CLN) IGHJ usage biased toward IGHJ4 in CNS and CLNs | • Class switched, acquired SHM and expanded clones in CNS B cells | • Both less mature and more experienced offspring observed in the CNS and CLN • Maturation steps not restricted to a single compartment • Clonal expansion of B cells possibly occurs in multiple compartments |
| (Johansen et al., 2015) | MS/n = 10 OIND/n = 6 | CSF/blood | NGS | IGH-V4 | • Higher frequency of IGHV4 genes and more replacement mutations in CSF of MS patients | Dominant B-cell clones • produce CSF IgG • are present at both sides of BBB • Go through several rounds of mutations in the CSF |
| (Eggers et al., 2017) | MS = 11 (bulk NGS) | CSF/blood | NGS | N/A | • ASC are clonally expanded in the CSF • ASC participate in production of clonal CSF IgG | • Clonal relationships between CSF and PB B cells • Migration of B cells and activation in the CNS in active MS • Clonal relationships between CSF and PB suggest influx of functionally diverse B cells |
| (Greenfield et al., 2019) | RRMS = 8 PPMS = 2 | CSF/blood | NGS | N/A | • Exclusive CSF IgG-VH clusters (10/10 patients) • Exclusive CSF IgM-VH (9/10) • mixed IgM and IgG clusters (5/10) | • Clonal connections between PB and CSF in 10/10 patients |
| (Kowarik et al., 2020) | MS = 8 | CSF/blood | NGS | VH4 (at baseline) | • In CSF 80% of VH sequences within clonal populations at baseline | Lineage analyses of clonal groups: • Bidirectional exchange across the BBB • M, DN and plasmablasts from PB contribute 30% to clonal groups emanating from PB, naïve B cells < 10% |
| Source | Patient | Treatment between T1 + T2 | Time between T1 + T2 (Months) | Overlapping CSF-Clones T1–T2 |
|---|---|---|---|---|
| Tomescu-Baciu et al., 2019 | MS1 | Natalizumab | 18 | 37 |
| MS2 | Interferon-beta 1a | 18 | 24 | |
| Greenfield et al., 2019 | 1 | Fingolimod | 14 | 8 |
| 2 | Interferon | 15 | 9 | |
| 3 | Dimethyl fumarate | 22 | 1 | |
| 4 | Natlizumab | 13 | 2 | |
| 5 | - | 12 | 1 | |
| 6 | Interferon | 13 | - | |
| 7 | Fingolimod | 15 | - | |
| 8 | Natalizumab | 9 | - | |
| 9 | - | 15 | - | |
| 10 | - | 13 | - | |
| Kowarik et al., 2020 | S1 | Natalizumab | 6 | 1 |
| S5 | Natalizumab | 6 | 5 | |
| S6 | Natalizumab | 6 | 5 | |
| S7 | Natalizumab | 6 | 0 | |
| S2 | Fingolimod | 6 | 0 | |
| S3 | Fingolimod | 6 | - | |
| S4 | Fingolimod | 6 | 0 | |
| S8 | Fingolimod | 6 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruschil, C.; Kemmerer, C.L.; Beller, L.; Gabernet, G.; Kowarik, M.C. Next Generation Sequencing of Cerebrospinal Fluid B Cell Repertoires in Multiple Sclerosis and Other Neuro-Inflammatory Diseases—A Comprehensive Review. Diagnostics 2021, 11, 1871. https://doi.org/10.3390/diagnostics11101871
Ruschil C, Kemmerer CL, Beller L, Gabernet G, Kowarik MC. Next Generation Sequencing of Cerebrospinal Fluid B Cell Repertoires in Multiple Sclerosis and Other Neuro-Inflammatory Diseases—A Comprehensive Review. Diagnostics. 2021; 11(10):1871. https://doi.org/10.3390/diagnostics11101871
Chicago/Turabian StyleRuschil, Christoph, Constanze Louisa Kemmerer, Lena Beller, Gisela Gabernet, and Markus Christian Kowarik. 2021. "Next Generation Sequencing of Cerebrospinal Fluid B Cell Repertoires in Multiple Sclerosis and Other Neuro-Inflammatory Diseases—A Comprehensive Review" Diagnostics 11, no. 10: 1871. https://doi.org/10.3390/diagnostics11101871
APA StyleRuschil, C., Kemmerer, C. L., Beller, L., Gabernet, G., & Kowarik, M. C. (2021). Next Generation Sequencing of Cerebrospinal Fluid B Cell Repertoires in Multiple Sclerosis and Other Neuro-Inflammatory Diseases—A Comprehensive Review. Diagnostics, 11(10), 1871. https://doi.org/10.3390/diagnostics11101871