
Characteristics and Predictors of Progression Interstitial Lung Disease in Rheumatoid Arthritis Compared with Other Autoimmune Disease: A Retrospective Cohort Study

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design
2.2. Study Population
2.3. Protocol
2.4. Working Definitions and Variables
2.5. Statistical Analysis
3. Results
3.1. Clinical and Epidemiological Characteristics
3.2. Clinical Course
3.3. Factors Associated with Progression of Lung Disease and Death in ILD-SAI
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robles-Pérez, A.; Luburich, P.; Bolivar, S.; Dorca, J.; Nolla, J.M.; Molina-Molina, M.; Narváez, J.A. A prospective study of lung disease in a cohort of early rheumatoid arthritis patients. Sci. Rep. 2020, 10, 15640. [Google Scholar] [CrossRef]
- Demoruelle, M.K.; Mittoo, S.; Solomon, J.J. Connective tissue disease-related interstitial lung disease. Best Pract. Res. Clin. Rheumatol. 2016, 30, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.D.; Medsger, T.A. Changes in causes of death in systemic sclerosis, 1972–2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottin, V.; Thivolet-Béjui, F.; Reynaud-Gaubert, M.; Cadranel, J.; Delaval, P.; Ternamian, P.J.; Cordier, J.F.; Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur. Respir. J. 2003, 22, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.; Ryerson, C.J.; Dunne, J.V.; Wilcox, P.G. Demographic and clinical predictors of progression and mortality in connective tissue disease-associated interstitial lung disease: A retrospective cohort study. BMC Pulm. Med. 2019, 19, 192. [Google Scholar] [CrossRef] [PubMed]
- Mena-Vázquez, N.; Godoy-Navarrete, F.J.; Manrique-Arija, S.; Aguilar-Hurtado, M.C.; Romero-Barco, C.M.; Ureña-Garnica, I.; Espildora, F.; Añón-Oñate, I.; Pérez-Albaladejo, L.; Gomez-Cano, C.; et al. Non-anti-TNF biologic agents are associated with slower worsening of interstitial lung disease secondary to rheumatoid arthritis. Clin. Rheumatol. 2020, 40, 133–142. [Google Scholar] [CrossRef]
- Kim, D.; Cho, S.K.; Choi, C.B.; Choe, J.Y.; Chung, W.T.; Hong, S.J.; Jun, J.B.; Jung, Y.O.; Kim, T.H.; Kim, T.J.; et al. Impact of interstitial lung disease on mortality of patients with rheumatoid arthritis. Rheumatol. Int. 2017, 37, 1735–1745. [Google Scholar] [CrossRef]
- Hyldgaard, C.; Hilberg, O.; Pedersen, A.B.; Ulrichsen, S.P.; Løkke, A.; Bendstrup, E.; Ellingsen, T. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: Comorbidity and mortality. Ann. Rheum. Dis. 2017, 76, 1700–1706. [Google Scholar] [CrossRef]
- Koduri, G.; Norton, S.; Young, A.; Cox, N.; Davies, P.; Devlin, J.; Dixey, J.; Gough, A.; Prouse, P.; Winfield, J.; et al. ERAS (Early Rheumatoid Arthritis Study). Interstitial lung disease has a poor prognosis in rheumatoid arthritis: Results from an inception cohort. Rheumatology 2010, 49, 1483–1489. [Google Scholar] [CrossRef] [Green Version]
- Kakutani, T.; Hashimoto, A.; Tominaga, A.; Kodama, K.; Nogi, S.; Tsuno, H.; Ogihara, H.; Nunokawa, T.; Komiya, A.; Furukawa, H.; et al. Related factors, increased mortality and causes of death in patients with rheumatoid arthritis-associated interstitial lung disease. Mod. Rheumatol. 2020, 30, 458–464. [Google Scholar] [CrossRef]
- Solomon, J.J.; Ryu, J.H.; Tazelaar, H.D.; Myers, J.L.; Tuder, R.; Cool, C.D.; Curran-Everett, D.; Fischer, A.; Swigris, J.J.; Brown, K.K. Fibrosing interstitial pneumonia predicts survival in patients with rheumatoid arthritis-associated interstitial lung disease (RA-ILD). Respir. Med. 2013, 107, 1247–1252. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.A.; Lee, J.S.; Park, J.K.; Lee, E.B.; Song, Y.W.; Lee, E.Y. Clinical characteristics associated with occurrence and poor prognosis of interstitial lung disease in rheumatoid arthritis. Korean. J. Intern. Med. 2019, 34, 434–441. [Google Scholar] [CrossRef]
- Kim, E.J.; Elicker, B.M.; Maldonado, F.; Webb, W.R.; Ryu, J.H.; Van Uden, J.H.; Lee, J.S.; King, T.E., Jr.; Collard, H.R. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur. Respir. J. 2010, 35, 1322–1328. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.C.; Choi, K.H.; Jacob, J.; Song, J.W. Prognostic role of blood KL-6 in rheumatoid arthritis-associated interstitial lung disease. PLoS ONE 2020, 15, e0229997. [Google Scholar] [CrossRef]
- Winstone, T.A.; Assayag, D.; Wilcox, P.G.; Dunne, J.V.; Hague, C.J.; Leipsic, J.; Collard, H.R.; Ryerson, C.J. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: A systematic review. Chest 2014, 146, 422–436. [Google Scholar] [CrossRef]
- Singh, N.; Varghese, J.; England, B.R.; Solomon, J.J.; Michaud, K.; Mikuls, T.R.; Healy, H.S.; Kimpston, E.M.; Schweizer, M.L. Impact of the pattern of interstitial lung disease on mortality in rheumatoid arthritis: A systematic literature review and meta-analysis. Semin. Arthritis Rheum. 2019, 49, 358–365. [Google Scholar] [CrossRef]
- Zamora-Legoff, J.A.; Krause, M.L.; Crowson, C.S.; Ryu, J.H.; Matteson, E.L. Patterns of interstitial lung disease and mortality in rheumatoid arthritis. Rheumatology 2017, 56, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Wang, L.; Li, L.; Li, Y.; Liu, R.; Zheng, Y. Risk factors for progression and prognosis of rheumatoid arthritis-associated interstitial lung disease: Single center study with a large sample of Chinese population. Clin. Rheumatol. 2019, 38, 1109–1116. [Google Scholar] [CrossRef]
- Hozumi, H.; Nakamura, Y.; Johkoh, T.; Sumikawa, H.; Colby, T.V.; Kono, M.; Hashimoto, D.; Enomoto, N.; Fujisawa, T.; Inui, N.; et al. Acute exacerbation in rheumatoid arthritis-associated interstitial lung disease: A retrospective case control study. BMJ Open 2013, 3, e003132. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Song, J.W.; Yoon, H.Y.; Cross, G.; Barnett, J.; Woo, W.L.; Adams, F.; Kokosi, M.; Devaraj, A.; Renzoni, E.; et al. Prevalence and Effects of Emphysema in Never-Smokers with Rheumatoid Arthritis Interstitial Lung Disease. EBioMedicine 2018, 28, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Kim, D.S.; Yoo, B.; Seo, J.B.; Rho, J.Y.; Colby, T.V.; Kitaichi, M. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest 2005, 127, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurmi, H.M.; Purokivi, M.K.; Kärkkäinen, M.S.; Kettunen, H.-P.; Selander, T.A.; Kaarteenaho, R.L. Variable course of disease of rheumatoid arthritis-associated usual interstitial pneumonia compared to other subtypes. BMC Pulm. Med. 2016, 16, 107. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Serrano, J.; Herrera-Bringas, D.; Pérez-Román, D.I.; Pérez-Dorame, R.; Mateos-Toledo, H.; Mejía, M. Rheumatoid arthritis-related interstitial lung disease (RA-ILD): Methotrexate and the severity of lung disease are associated to prognosis. Clin. Rheumatol. 2017, 36, 1493–1500. [Google Scholar] [CrossRef]
- Solomon, J.J.; Chung, J.H.; Cosgrove, G.P.; Demoruelle, M.K.; Fernandez-Perez, E.R.; Fischer, A.; Frankel, S.K.; Hobbs, S.B.; Huie, T.J.; Ketzer, J.; et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur. Respir. J. 2016, 47, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Takayanagi, N.; Sugiura, H.; Miyahara, Y.; Tokunaga, D.; Kawabata, Y.; Sugita, Y. Lung diseases directly associated with rheumatoid arthritis and their relationship to outcome. Eur. Respir. J. 2011, 37, 1411–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morisset, J.; Vittinghoff, E.; Lee, B.Y.; Tonelli, R.; Hu, X.; Elicker, B.M.; Ryu, J.H.; Jones, K.D.; Cerri, S.; Manfredi, A.; et al. The performance of the GAP model in patients with rheumatoid arthritis associated interstitial lung disease. Respir. Med. 2017, 127, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Distler, O.; Brown, K.K.; Distler, J.H.W.; Assassi, S.; Maher, T.M.; Cottin, V.; Varga, J.; Coeck, C.; Gahlemann, M.; Sauter, W.; et al. Design of a randomised, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENSCIS™). Clin. Exp. Rheumatol. 2017, 35 (Suppl. 106), 75–81. [Google Scholar]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Narváez, J.; Robles-Pérez, A.; Molina-Molina, M.; Vicens-Zygmunt, V.; Luburich, P.; Yañez, M.A.; Alegre, J.J.; Nolla, J.M. Real-world clinical effectiveness of rituximab rescue therapy in patients with progressive rheumatoid arthritis-related interstitial lung disease. Semin. Arthritis Rheum. 2020, 50, 902–910. [Google Scholar] [CrossRef]
- Md Yusof, M.Y.; Kabia, A.; Darby, M.; Lettieri, G.; Beirne, P.; Vital, E.M.; Dass, S.; Emery, P. Effect of rituximab on the progression of rheumatoid arthritis-related interstitial lung disease: 10 years’ experience at a single centre. Rheumatology 2017, 56, 1348–1357. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Díaz, C.; Loricera, J.; Castañeda, S.; López-Mejías, R.; Ojeda-García, C.; Olivé, A.; Rodríguez-Muguruza, S.; Carreira, P.E.; Pérez-Sandoval, T.; Retuerto, M.; et al. Abatacept in patients with rheumatoid arthritis and interstitial lung disease: A national multicenter study of 63 patients. Semin. Arthritis Rheum. 2018, 48, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Díaz, C.; Castañeda, S.; Melero-González, R.B.; Ortiz-Sanjuán, F.; Juan-Mas, A.; Carrasco-Cubero, C.; Casafont-Solé, I.; Olivé, A.; Rodríguez-Muguruza, S.; Almodóvar-González, R.; et al. Abatacept in interstitial lung disease associated with rheumatoid arthritis: National multicenter study of 263 patients. Rheumatology 2020, 59, 3906–3916. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.; Cassone, G.; Furini, F.; Gremese, E.; Venerito, V.; Atzeni, F.; Arrigoni, E.; Della Casa, G.; Cerri, S.; Govoni, M.; et al. Tocilizumab therapy in rheumatoid arthritis with interstitial lung disease: A multicenter retrospective study. Intern. Med. J. 2019, 50, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Bosello, S.L.; De Luca, G.; Rucco, M.; Berardi, G.; Falcione, M.; Danza, F.M.; Pirronti, T.; Ferraccioli, G. Long-term efficacy of B cell depletion therapy on lung and skin involvement in diffuse systemic sclerosis. Semin. Arthritis Rheum. 2015, 44, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Dominique, S.; Janvresse, A.; Levesque, H.; Menard, J.F. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir. Med. 2012, 106, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Bohan, A.; Peter, J.B. Polymyositis and dermatomyositis (first of two parts). N. Engl. J. Med. 1975, 292, 344–347. [Google Scholar] [CrossRef]
- Bohan, A.; Peter, J.B. Polymyositis and dermatomyositis (second of two parts). N. Engl. J. Med. 1975, 292, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A., Jr.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Su, R.; Bennett, M.; Jacobs, S.; Hunter, T.; Bailey, C.; Krishnan, E.; Rosen, G.; Chung, L. An analysis of connective tissue disease-associated interstitial lung disease at a US Tertiary Care Center: Better survival in patients with systemic sclerosis. J. Rheumatol. 2011, 38, 693–701. [Google Scholar] [CrossRef]
- Jacob, J.; Bartholmai, B.J.; Rajagopalan, S.; Brun, A.L.; Egashira, R.; Karwoski, R.; Kokosi, M.; Wells, A.U.; Hansell, D.M. Evaluation of computer-based computer tomography stratification against outcome models in connective tissue disease-related interstitial lung disease: A patient outcome study. BMC Med. 2016, 14, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Kim, D.S.; Park, I.N.; Jang, S.J.; Kitaichi, M.; Nicholson, A.G.; Colby, T.V. Prognosis of fibrotic interstitial pneumonia: Idiopathic versus collagen vascular disease-related subtypes. Am. J. Respir. Crit. Care Med. 2007, 175, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.A.; Nisar, M.; Arthanari, S.; Carty, S.; Woodhead, F.A.; Price-Forbes, A.; Middleton, D.; Dempsey, O.; Miller, D.; Basu, N.; et al. Rheumatoid arthritis related interstitial lung disease-improving outcomes over 25 years: A large multicentre UK study. Rheumatology 2020, 60, 1882–1890. [Google Scholar] [CrossRef]
- Mena-Vázquez, N.; Rojas-Gimenez, M.; Romero-Barco, C.M.; Manrique-Arija, S.; Francisco, E.; Aguilar-Hurtado, M.C.; Añón-Oñate, I.; Pérez-Albaladejo, L.; Ortega-Castro, R.; Godoy-Navarrete, F.J.; et al. Predictors of Progression and Mortality in Patients with Prevalent Rheumatoid Arthritis and Interstitial Lung Disease: A Prospective Cohort Study. J. Clin. Med. 2021, 10, 874. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.; Vummidi, D.; Khanna, D. Management of connective tissue diseases associated interstitial lung disease: A review of the published literature. Curr. Opin. Rheumatol. 2016, 28, 236–245. [Google Scholar] [CrossRef]
- Navaratnam, V.; Ali, N.; Smith, C.J.; McKeever, T.; Fogarty, A.; Hubbard, R.B. Does the presence of connective tissue disease modify survival in patients with pulmonary fibrosis? Respir. Med. 2011, 105, 1925–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moua, T.; Zamora Martinez, A.C.; Baqir, M.; Vassallo, R.; Limper, A.H.; Ryu, J.H. Predictors of diagnosis and survival in idiopathic pulmonary fibrosis and connective tissue disease-related usual interstitial pneumonia. Respir. Res. 2014, 15, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurmi, H.M.; Purokivi, M.K.; Kärkkäinen, M.S.; Kettunen, H.P.; Selander, T.A.; Kaarteenaho, R.L. Are risk predicting models useful for estimating survival of patients with rheumatoid arthritis-associated interstitial lung disease? BMC Pulm. Med. 2017, 17, 16. [Google Scholar] [CrossRef] [Green Version]
- Yıldırım, F.; Türk, M.; Bitik, B.; Erbaş, G.; Köktürk, N.; Haznedaroğlu, Ş.; Türktaş, H. Comparison of clinical courses and mortality of connective tissue disease-associated interstitial pneumonias and chronic fibrosing idiopathic interstitial pneumonias. Kaohsiung J. Med. Sci. 2019, 35, 365–372. [Google Scholar] [CrossRef]
- Bouros, D.; Wells, A.U.; Nicholson, A.G.; Colby, T.V.; Polychronopoulos, V.; Pantelidis, P.; Haslam, P.L.; Vassilakis, D.A.; Black, C.M.; du Bois, R.M. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am. J. Respir. Crit. Care Med. 2002, 165, 1581–1586. [Google Scholar] [CrossRef]
- Reiseter, S.; Gunnarsson, R.; Mogens Aaløkken, T.; Lund, M.B.; Mynarek, G.; Corander, J.; Haydon, J.; Molberg, Ø. Progression and mortality of interstitial lung disease in mixed connective tissue disease: A long-term observational nationwide cohort study. Rheumatology 2018, 57, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Sharp, C.; McCabe, M.; Dodds, N.; Edey, A.; Mayers, L.; Adamali, H.; Millar, A.B.; Gunawardena, H. Rituximab in autoimmune connective tissue disease-associated interstitial lung disease. Rheumatology 2016, 55, 1318–1324. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.J.; Brown, K.K. Rheumatoid arthritis-associated interstitial lung disease. Open Access Rheumatol. 2012, 4, 21–31. [Google Scholar] [PubMed] [Green Version]
- McNearney, T.A.; Reveille, J.D.; Fischbach, M.; Friedman, A.W.; Lisse, J.R.; Goel, N.; Tan, F.K.; Zhou, X.; Ahn, C.; Feghali-Bostwick, C.A.; et al. Pulmonary involvement in systemic sclerosis: Associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum. 2007, 57, 318–326. [Google Scholar] [CrossRef] [PubMed]
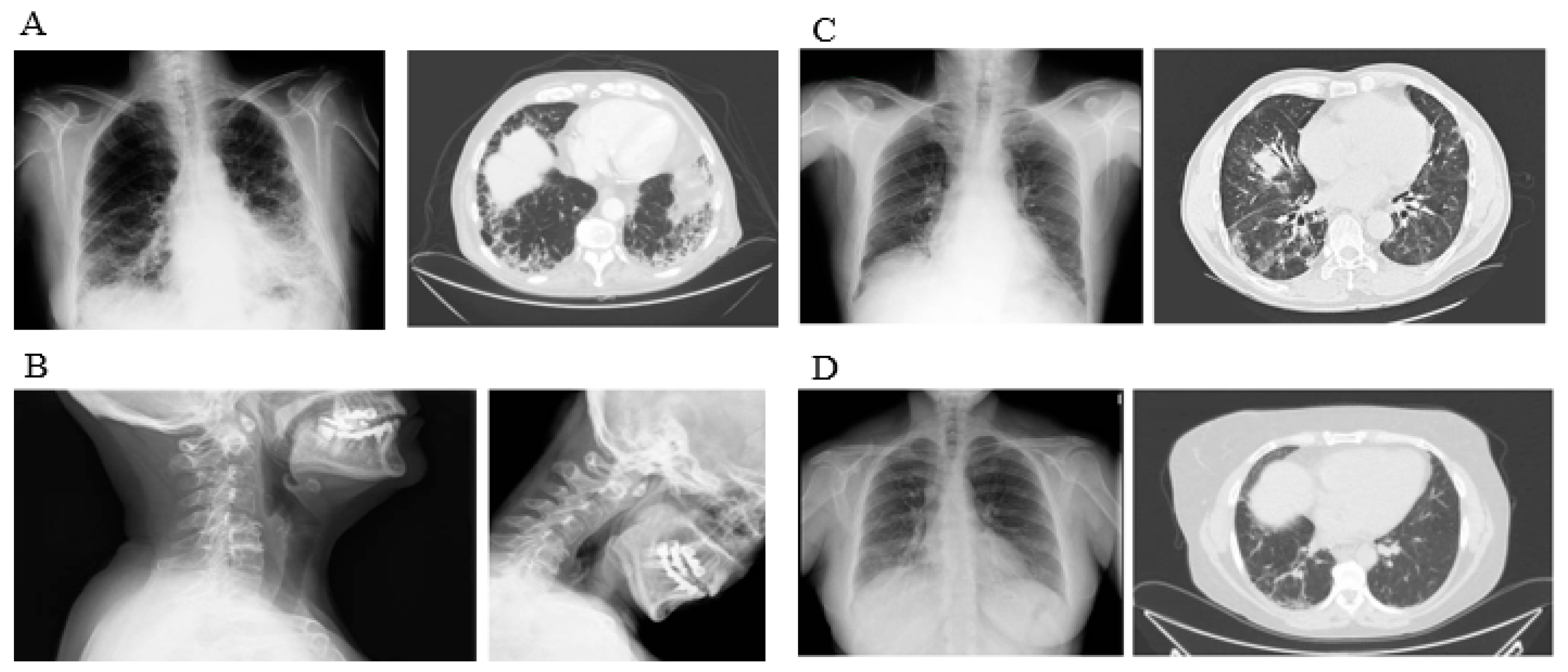

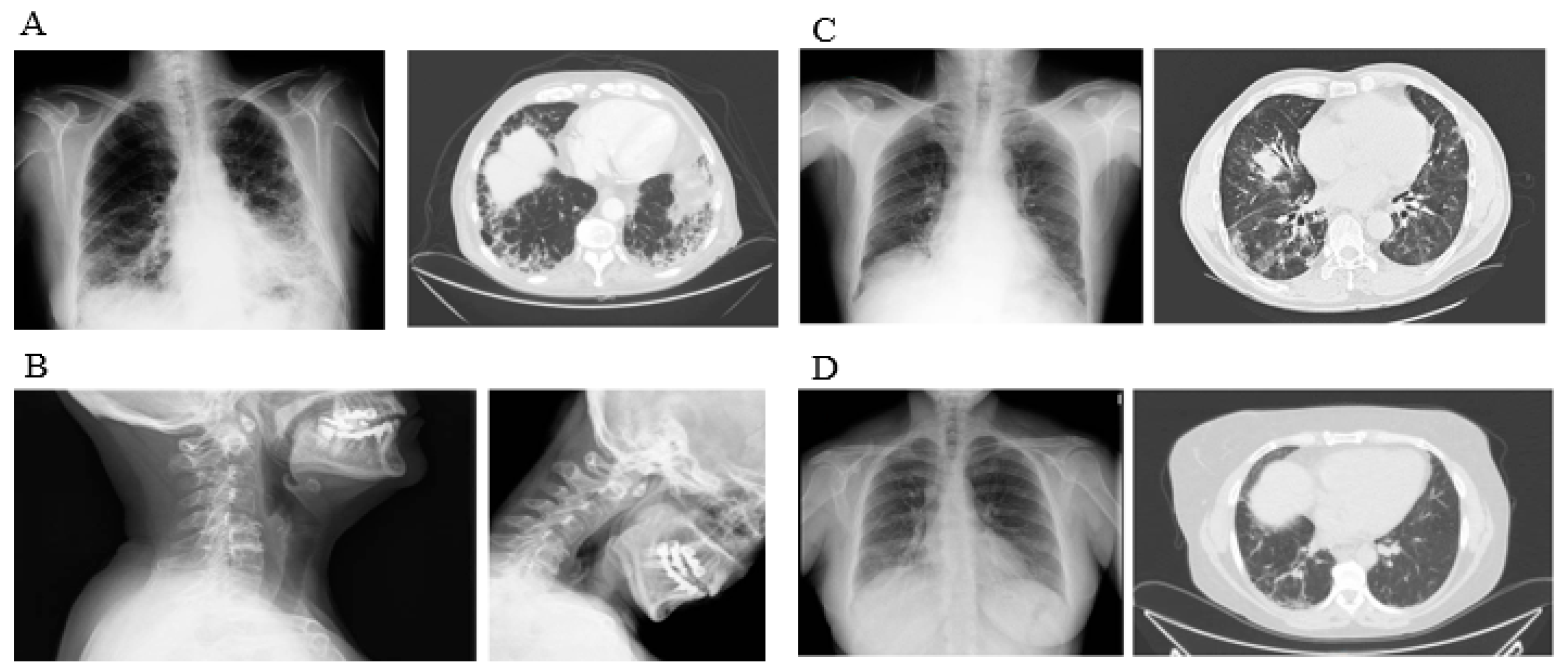{kind=link}
{kind=link}
| Total (n = 204) | RA (n = 123) | IM (n = 23) | SS (n = 58) | p Value | |
|---|---|---|---|---|---|
| Epidemiological characteristics | |||||
| Female sex, n (%) | 136 (66.7) | 67 (54.5) | 18 (78.3) | 51 (87.9) | <0.001 |
| Caucasian race, n (%) | 198 (97.1) | 119 (96.7) | 23 (100) | 56 (96.6) | 0.673 |
| Age, years, mean (SD) | 65.3 (12.6) | 69.2 (9.6) | 55.5 (18.5) | 61.0 (12.2) | <0.001 |
| Clinical and laboratory characteristics | |||||
| Smoking | 0.002 | ||||
| Nonsmokers, n (%) | 112 (54.9) | 54 (43.9) | 19 (82.6) | 39 (67.2) | |
| Smokers, n (%) | 36 (17.6) | 28 (22.8) | 1 (4.3) | 7 (12.1) | |
| Exsmokers, n (%) | 56 (27.5) | 41 (33.3) | 3 (13.0) | 12 (20.7) | |
| Time with SAI, months, median (IQR) | 124.0 (57.4–209.7) | 146.3 (68.2–218.1) | 51.7 (25.1–157.5) | 119.3 (56.6–206.1) | 0.037 |
| Time with ILD, months, median (IQR) | 56.5 (29.8–93.3) | 51.8 (28.5–85.9) | 33.9 (25.1–80.5) | 65.5 (40.3–140.1) | 0.059 |
| RF-positive (>10), n (%) | 122 (59.8) | 119 (96.7) | 1 (4.3) | 2 (3.4) | <0.001 |
| ACPA (>20), n (%) | 107 (52.5) | 107 (87.0) | 0 (0.0) | 0 (0.0) | <0.001 |
| ANA-positive, n (%) | 104 (51.0) | 30 (24.6) | 19 (82.6) | 55 (94.8) | <0.001 |
| Anti-SCL70, n (%) | 28 (13.8) | 0 (0.0) | 0 (0.0) | 28 (48.3) | <0.001 |
| Anticentromere, n (5) | 20 (9.8) | 0 (0.0) | 0 (0.0) | 20 (34.4) | <0.001 |
| PM-SCL, n (%) | 3 (1.5) | 0 (0.0) | 0 (0.0) | 3 (5.1) | 0.033 |
| RNP, n (%) | 2 (1.0) | 0 (0.0) | 0 (0.0) | 2 (3.4) | 0.079 |
| Anti-Ku, n (%) | 1 (0.5) | 0 (0.0) | 0 (0.0) | 2 (3.4) | 0.079 |
| RNA-polymerase 3, n (%) | 1 (0.5) | 0 (0.0) | 0 (0.0) | 1 (1.7) | 0.282 |
| Anti-Jo, n (%) | 6 (2.9) | 0 (0.0) | 6 (26.1) | 0 (0.0) | <0.001 |
| Anti-PL7, n (%) | 5 (2.5) | 0 (0.0) | 5 (21.7) | 0 (0.0) | <0.001 |
| Anti-EJ, n (%) | 1 (0.5) | 0 (0.0) | 1 (4.3) | 0 (0.0) | 0.019 |
| Anti-NMDA, n (%) | 3 (1.5) | 0 (0.0) | 3 (13.0) | 0 (0.0) | <0.001 |
| Anti-TIF, n (%) | 2 (1.0) | 0 (0.0) | 2 (8.7) | 0 (0.0) | <0.001 |
| Anti-SRP, n (%) | 1 (0.5) | 0 (0.0) | 1 (4.3) | 0 (0.0) | 0.019 |
| Secondary Sjögren syndrome, n (%) | 33 (16.2) | 18 (14.6) | 6 (26.1) | 9 (15.5) | 0.387 |
| Treatment | |||||
| Conventional synthetic DMARDs, n (%) | 129 (63.5) | 105 (85.4) | 12 (52.2) | 12 (20.7) | <0.001 |
| Methotrexate, n (%) | 62 (30.4) | 53 (43.1) | 4 (17.4) | 5 (8.6) | <0.001 |
| Leflunomide, n (%) | 32 (15.7) | 31 (25.2) | 0 (0.0) | 1 (1.7) | <0.001 |
| Sulfasalazine, n (%) | 9 (4.4) | 9 (7.3) | 0 (0.0) | 0 (0.0) | <0.001 |
| Hydroxychloroquine, n (%) | 36 (17.7) | 23 (18.7) | 8 (34.7) | 5 (8.6) | 0.016 |
| Biologic DMARDs, n (%) | 78 (38.2) | 57 (46.3) | 6 (26.1) | 15 (25.9) | 0.013 |
| Anti-TNF, n (%) | 14 (6.9) | 14 (11.4) | 0 (0.0) | 0 (0.0) | 0.343 |
| Tocilizumab, n (%) | 8 (6.9) | 5 (4.1) | 1 (4.3) | 2 (3.4) | 0.343 |
| Abatacept, n (%) | 19 (9.3) | 19 (15.4) | 0 (0.0) | 0 (0.0) | 0.001 |
| Rituximab, n (%) | 37 (18.1) | 19 (15.4) | 4 (17.4) | 14 (24.1) | 0.365 |
| Immunosuppressants | 75 (36.9) | 15 (12.2) | 20 (87.0) | 40 (68.9) | <0.001 |
| Mycophenolate, n (%) | 58 (28.5) | 10 (8.1) | 13 (56.5) | 35 (60.3) | <0.001 |
| Azathioprine, n (%) | 14 (6.9) | 5 (4.1) | 6 (26.1) | 3 (5.1) | 0.009 |
| Cyclophosphamide, n (%) | 3 (1.5) | 0 (0.0) | 1 (4.3) | 2 (3.4) | 0.095 |
| Antifibrotic drugs, nintedanib n (%) | 4 (2.0) | 2 (1.6) | 0 (0.0) | 2 (3.4) | 0.549 |
| Corticosteroids, n (%) | 134 (66.0) | 88 (71.5) | 19 (82.6) | 27 (46.6) | 0.004 |
| Dose of corticosteroids, median (IQR) | 5.0 (0.0–7.5) | 5.0 (0.0–7.5) | 5.0 (5.0–10.0) | 2.5 (0.0–5.0) | 0.003 |
| Total (n = 204) | RA (n = 123) | IM (n = 23) | SS (n = 58) | p Value | |
|---|---|---|---|---|---|
| Infections, n (%) | 132 (64.7) | 82 (66.7) | 15 (65.2) | 35 (60.3) | 0.666 |
| Respiratory infection, n (%) | 116 (56.9) | 76 (61.8) | 12 (52.2) | 28 (48.3) | 0.205 |
| Other infections, n (%) | 59 (28.9) | 34 (27.6) | 7 (30.4) | 18 (31.0) | 0.883 |
| Cold sore (herpes), n (%) | 10 (4.9) | 7 (5.6) | 1 (4.3) | 2 (3.4) | 0.242 |
| Cutaneous, n (%) | 25 (12.2) | 9 (7.3) | 4 (17.3) | 12 (20.6) | 0.032 |
| Urinary infection, n (%) | 28 (13.7) | 19 (15.4) | 3 (13.0) | 6 (10.3) | 0.460 |
| Hospitalization, n (%) | 75 (36.8) | 55 (44.7) | 4 (17.4) | 16 (27.6) | 0.010 |
| Reason for hospitalization | 0.032 | ||||
| Respiratory infection, n (%) | 48 (23.5) | 37 (30.1) | 4 (17.4) | 7 (12.1) | |
| Progression of ILD, n (%) | 21 (10.3) | 15 (12.2) | 0 (0.0) | 6 (10.3) | |
| Other causes, n (%) | 6 (2.9) | 3 (2.4) | 0 (0.0) | 3 (5.2) | |
| Mortality, n (%) | 29 (14.2) | 19 (15.4) | 1 (4.3) | 9 (15.5) | 0.355 |
| Total (n = 204) | RA (n = 123) | IM (n = 23) | SS (n = 58) | p Value | ||
|---|---|---|---|---|---|---|
| Outcomes clinical course ** | 0.016 | |||||
| Improvement, n (%) | Final | 33 (16.1) | 14 (11.4) | 9 (39.1) | 10 (17.2) | |
| Stabilization, n (%) | Final | 98 (48.0) | 61 (49.5) | 10 (43.5) | 27 (46.6) | |
| Worsening, n (%) | Final | 44 (21.5) | 29 (23.5) | 3 (13.0) | 12 (20.7) | |
| Death, n (%) | Final | 29 (14.2) | 19 (15.4) | 1 (4.3) | 9 (15.5) | |
| Pulmonary function testing | ||||||
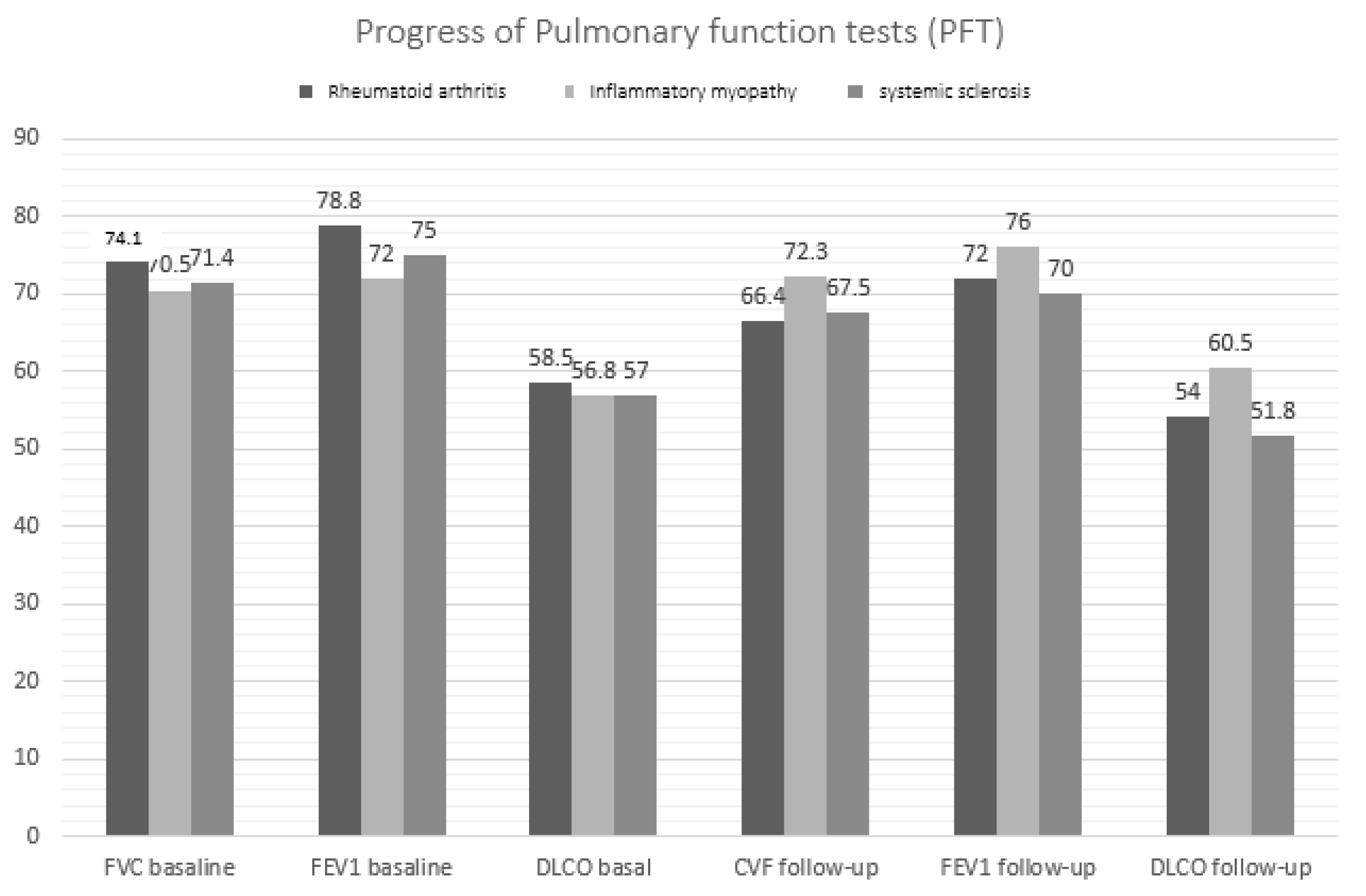
| FVC, mean (SD) | Baseline | 72.9 (16.6) | 74.1 (15.8) | 70.5 (15.8) | 71.4 (18.5) | 0.465 |
| Final | 68.2 (16.2) * | 66.4 (21.4) * | 72.3 (15.8) | 67.5 (22.8) * | 0.036 | |
| FVC < 80%, n (%) | Baseline | 119 (58.3) | 73 (60.9) | 11 (49.8) | 35 (60.3) | 0.772 |
| Final | 134 (65.6) * | 83 (67.4) * | 13 (56.5) | 38 (65.5) * | 0.045 | |
| FEV1, mean (SD) | Baseline | 77.7 (16.2) | 78.8 (16.2) | 72.0 (15.5) | 75.5 (15.9) | 0.168 |
| Final | 72.9 (19.6) * | 71.9 (20.4) * | 76.0 (17.1) | 70.0 (18.5) | 0.240 | |
| DLCO-SB, mean (SD) | Baseline | 57.8 (15.0) | 58.5 (15.0) | 56.8 (14.3) | 56.9 (15.4) | 0.775 |
| Final | 53.8 (16.4) * | 54.1 (16.2) * | 60.5 (15.1) | 51.8 (16.5) * | 0.026 | |
| HRCT pattern | ||||||
| Radiologic pattern | <0.001 | |||||
| UIP, n (%) | Baseline | 86 (42.1) | 74 (60.1) | 2 (8.7) | 10 (17.2) | |
| Final | 91 (44.6) | 77 (62.6) | 2 (8.7) | 15 (25.8) | ||
| NSIP, n (%) | Baseline | 101 (49.5) | 35 (28.5) | 20 (86.9) | 46 (79.3) | |
| Final | 99 (48.5) | 33 (26.8) | 20 (86.9) | 42 (72.4) | ||
| Fibrotic NSIP, n (%) | Baseline | 10 (4.9) | 7 (5.6) | 1 (4.3) | 2 (3.4) | |
| Final | 7 (3.4) | 6 (4.8) | 1 (4.3) | 1 (3.4) | ||
| Other types, n (%) | Baseline | 7 (3.4) | 7 (5.6) | 0 (0.0) | 0 (0.0) | |
| Final | 7 (3.4) | 7 (5.6) | 0 (0.0) | 0 (0.0) | ||
| Progression by HRCT | 0.002 | |||||
| Progression, n (%) | Final | 72 (35.3) | 48 (39.0) | 4 (17.4) | 20 (34.5) | |
| Stabilization, n (%) | Final | 93 (45.6) | 60 (48.8) | 7 (30.4) | 26 (44.8) | |
| Improvement, n (%) | Final | 39 (19.1) | 15 (12.2) | 12 (52.2) | 12 (20.7) | |
| Variable | Univariate HR (95% CI) | Multivariate HR (95% CI) | p Value |
|---|---|---|---|
| Age, years | 1.025 (1.00–1.05) | ||
| Male sex | 1.172 (0.64–2.14) | ||
| Current or previous smoking | 2.419 (1.48–3.94) | 2.799 (1.64–4.75) | 0.010 |
| UIP pattern | 2.317 (1.44–3.71) | 1.787 (1.06–2.99) | 0.028 |
| Positive ACPA or scl70 values | 2.046 (1.31–3.19) | ||
| Baseline FVC < 80% | 2.840 (1.47–5.48) | 2.348 (1.40–3.92) | 0.015 |
| Baseline DLCO-SB < 80% | 3.696 (0.86–9.12) | ||
| Corticosteroids | 1.434 (0.76–2.08) | ||
| csDMARDs | 1.080 (0.595–1.96) | ||
| Immunosuppressants | 0.937 (0.52–1.66) | ||
| bDMARDs | 0.744 (0.40–1.35) | ||
| SAI subtype | 2.101 (1.20–3.66) | 1.901 (1.32–2.73) | 0.027 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mena-Vázquez, N.; Rojas-Gimenez, M.; Romero-Barco, C.M.; Manrique-Arija, S.; Hidalgo Conde, A.; Arnedo Díez de los Ríos, R.; Cabrera César, E.; Ortega-Castro, R.; Espildora, F.; Aguilar-Hurtado, M.C.; et al. Characteristics and Predictors of Progression Interstitial Lung Disease in Rheumatoid Arthritis Compared with Other Autoimmune Disease: A Retrospective Cohort Study. Diagnostics 2021, 11, 1794. https://doi.org/10.3390/diagnostics11101794
Mena-Vázquez N, Rojas-Gimenez M, Romero-Barco CM, Manrique-Arija S, Hidalgo Conde A, Arnedo Díez de los Ríos R, Cabrera César E, Ortega-Castro R, Espildora F, Aguilar-Hurtado MC, et al. Characteristics and Predictors of Progression Interstitial Lung Disease in Rheumatoid Arthritis Compared with Other Autoimmune Disease: A Retrospective Cohort Study. Diagnostics. 2021; 11(10):1794. https://doi.org/10.3390/diagnostics11101794
Chicago/Turabian StyleMena-Vázquez, Natalia, Marta Rojas-Gimenez, Carmen María Romero-Barco, Sara Manrique-Arija, Ana Hidalgo Conde, Rocío Arnedo Díez de los Ríos, Eva Cabrera César, Rafaela Ortega-Castro, Francisco Espildora, María Carmen Aguilar-Hurtado, and et al. 2021. "Characteristics and Predictors of Progression Interstitial Lung Disease in Rheumatoid Arthritis Compared with Other Autoimmune Disease: A Retrospective Cohort Study" Diagnostics 11, no. 10: 1794. https://doi.org/10.3390/diagnostics11101794
APA StyleMena-Vázquez, N., Rojas-Gimenez, M., Romero-Barco, C. M., Manrique-Arija, S., Hidalgo Conde, A., Arnedo Díez de los Ríos, R., Cabrera César, E., Ortega-Castro, R., Espildora, F., Aguilar-Hurtado, M. C., Añón-Oñate, I., Pérez-Albaladejo, L., Abarca-Costalago, M., Ureña-Garnica, I., Velloso-Feijoo, M. L., Redondo-Rodriguez, R., & Fernández-Nebro, A. (2021). Characteristics and Predictors of Progression Interstitial Lung Disease in Rheumatoid Arthritis Compared with Other Autoimmune Disease: A Retrospective Cohort Study. Diagnostics, 11(10), 1794. https://doi.org/10.3390/diagnostics11101794